Journal of Clinical Endocrinology and Metabolism
Vol. 82, No. 1, The Endocrine Society, 1997
Potassium Bicarbonate Reduces Urinary Nitrogen Excretion in
Postmenopausal Women
L. FRASSETTO, R. CURTIS MORRIS, JR., AND A. SEBASTIAN
Department of Medicine and General Clinical Research Center,
University of California, San Francisco, California 94143
ABSTRACT
Previously we demonstrated that low grade chronic metabolic
acidosis exists normally in humans eating ordinary diets that
yield normal net rates of endogenous acid production (EAP), and
that the degree of acidosis increases with age. We hypothesize
that such diet-dependent and age-amplifying low grade metabolic
acidosis contributes to the decline in skeletal muscle mass that
occurs normally with aging. This hypothesis is based on the
reported finding that chronic metabolic acidosis induces muscle
protein breakdown, and that correction of acidosis reverses the
effect. Accordingly, in 14 healthy postmenopausal women residing
in a General Clinical Research Center and eating a constant diet
yielding a normal EAP rate, we tested whether correcting their
“physiological” acidosis with orally administered
potassium bicarbonate (KHCO3; 60-120 mmol/day for 18
days) reduces their urinary nitrogen loss. KHCO3
reduced EAP to nearly zero, significantly reduced the blood
hydrogen ion concentration (P < 0.001), and increased
the plasma bicarbonate concentration (P < 0.001),
indicating that pre-KHCO3 diet-dependent EAP was
significantly perturbing systemic acid-base equilibrium, causing
a low grade metabolic acidosis. Urinary ammonia nitrogen, urea
nitrogen, and total nitrogen levels significantly decreased. The
cumulative reduction in nitrogen excretion was 14.1 ± 12.3
g (P < 0.001). Renal creatinine clearance and urine
volume remained unchanged. We conclude that in postmenopausal
women, neutralization of diet-induced EAP with KHCO3
corrects their preexisting diet-dependent low grade metabolic
acidosis and significantly reduces their urinary nitrogen
wasting. The magnitude of the KHCO3-induced
nitrogen-sparing effect is potentially sufficient to both prevent
continuing age-related loss of muscle mass and restore previously
accrued deficits. (J Clin Endocrinol Metab
82: 254-259, 1997)
In disorders that cause chronic metabolic acidosis, protein
degradation in skeletal muscle is accelerated (1-3), increasing
the production of nitrogen (N) end products that are eliminated
in the urine, thereby inducing negative N balance (3). This
disturbance of N metabolism apparently results directly from the
acidosis, not from its cause or from other sequelae of the
underlying acidosis-producing disorder, because it occurs with
widely differing acidosis-producing conditions (2-7), and it is
reversible by administration of alkali (8-11), which corrects the
acidosis but not its cause. .Acidosis-induced proteolysis appears
to be an acid-base homeostatic mechanism. By releasing increased
amounts of amino acids, in particular glutamine, which is used by
the kidney for synthesis of ammonia, the kidney can increase the
excretion of acid (as ammonium) in the urine, thereby mitigating
the severity of the acidosis (2, 3, 12, 13).
One cause of chronic metabolic acidosis is eating a diet whose
metabolism yields noncarbonic acids (e.g. sulfuric acid) in
excess of base (e.g. bicarbonate) (14). Such diets induce a
chronic low grade metabolic acidosis even in healthy subjects;
the severity of the acidosis correlates with the net rate of
endogenous acid production (acid minus base) (14, 15). Net
acid-producing diets are typical of those ingested by the
inhabitants of industrialized countries (14, 15), where
consumption of foods rich in acid precursors (animal foods) is
disproportionate to that of foods rich in base precursors
(vegetable foods). Habitual ingestion of such diets has been
linked to clinical disorders, in particular nephrolithiasis and
osteoporosis, in which diet-induced acid production has been
identified as contributing to their pathogenesis (16-19).
Because of the attendant low grade chronic acidosis, habitual
ingestion of typical net acid-producing diets might chronically
sustain a slightly increased state of protein breakdown and
consequent N wasting. Conceivably, low grade N wasting is a tonic
“normal” state in adult humans, accounting for the
normal progressive decrease in muscle mass as adults get older
(20, 21). Indeed, diet-dependent acidosis induced muscle wasting
might be amplified by age because diet-dependent metabolic
acidosis tends to increase in severity with age (22, 23), which,
in turn, appears to result from the normal age-related decline in
the function of the kidney (24).
These considerations prompted us to investigate, in healthy
adult subjects eating a normal net acid-producing diet, whether
mitigation of the normal dietary net acid load influences urinary
N excretion.
Materials and Methods
In 14 healthy postmenopausal women residing in the University
of California-San Francisco General Clinical Research Center and
eating a constant net acid-producing diet, we tested the effect
of reducing the dietary net acid load on the rate of excretion of
N in the urine. We reduced the diet acid load without changing
the diet composition, by administering a dietary supplement of
the absorbable base, potassium bicarbonate (KHCO3).
The protocol was approved by the University of California-San
Francisco committee on human research, and informed consent was
obtained from each woman. The women ranged in age from 51-77 yr,
in weight from 53-76 kg, in height from 153-175 cm, and in body
mass index from 21-28 kg/m2. All were at least 5 yr
postmenopausal, and none was taking estrogen.
Each woman ate a constant diet (per 60 kg BW), containing 96
± 1 g protein (15.4 g N) and 1995 ± 17 Cal, which
yielded an endogenous acid production rate of 1.25 mEq/kg BW
(normal range, 0.5-1.5 mEq/kg BW). The diet was begun
approximately 2 weeks before the control period, before the
collection of specimens for measurements of plasma and urine
acid-base, electrolyte, and N composition was initiated, to
establish a steady state on the specific diet (adaptation
period). Light activity levels were maintained throughout the
study. After a subsequent 6-day control period, oral
KHCO3 was given as a dietary supplement at a dose of
60-120 mmol/day for 18 days (KHCO3 period). Balance
studies were continued for an additional 12 days after
KHCO3 was stopped (recovery period).
Sample collection
Arterialized venous blood samples were collected between
1530-1630 h, at least 3 h after the noon meal, without stasis or
exposure to air, from a vein on the back of the hand, which was
warmed in a water bath at 44 C for 5 min. Blood specimens were
obtained on 18 of 29 days throughout the study. The specimens
were analyzed for blood pH and carbon dioxide tension, plasma
total carbon dioxide content, blood urea, N, and serum
creatinine.
Each voided urine specimen was maintained under a thin layer
of mineral oil, preserved with thymol, and pooled in 24-h
collections for determination of pH and carbon dioxide content.
In addition, the total volume and the concentrations of ammonium,
titratable acid, creatinine, and urea were measured.
Analytical methods
Analytical methods for measuring blood and urine acid-base
parameters have been previously described (16, 23). Net acid
excretion was calculated as the sum of the excretion rates of
titratable acid and ammonium minus that of bicarbonate. The
titratable acid concentration was determined by titration, and
the urinary ammonium concentration was determined by the phenol
method. Blood urea N was determined by enzymatic reaction, and
blood and urinary creatinine were measured by the Jaffe method.
Total N was calculated as the sum of urea N, creatinine N, and
ammonium N.
Statistical analysis
For each blood and urine variable, the all-subject averages
were computed for each day and used as the primary database. The
results were analyzed by paired t tests and by linear,
nonlinear, and multiple; analysis using Sigmastat Jandel Co., San
Raphael, CA) and Statistica (Statsoft, Tulsa, OK). The results
are presented as the mean ± SD.
Results
During the control period before KHCO3
administration, all measured variables were stable, including the
urinary excretion rates of urea, creatinine, ammonia, total N,
and net acid; blood hydrogen ion ([H+]b)
and plasma bicarbonate concentrations ([HCO]p; Figs.
1-3); and serum urea and creatinine concentrations.
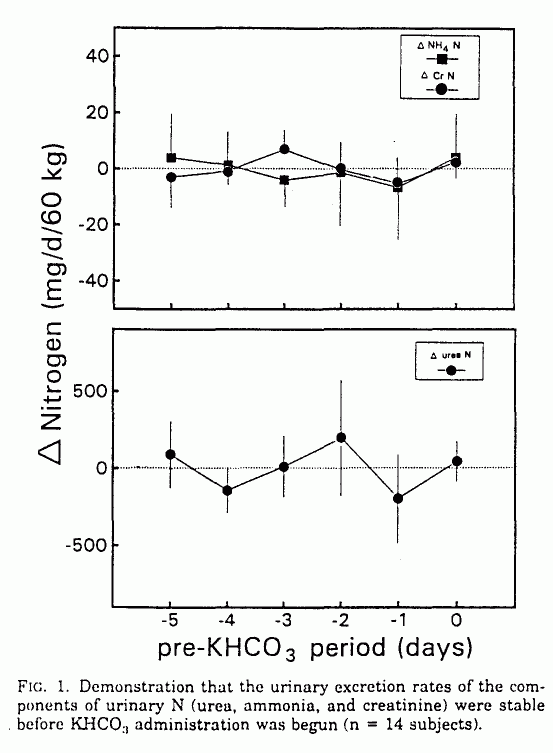
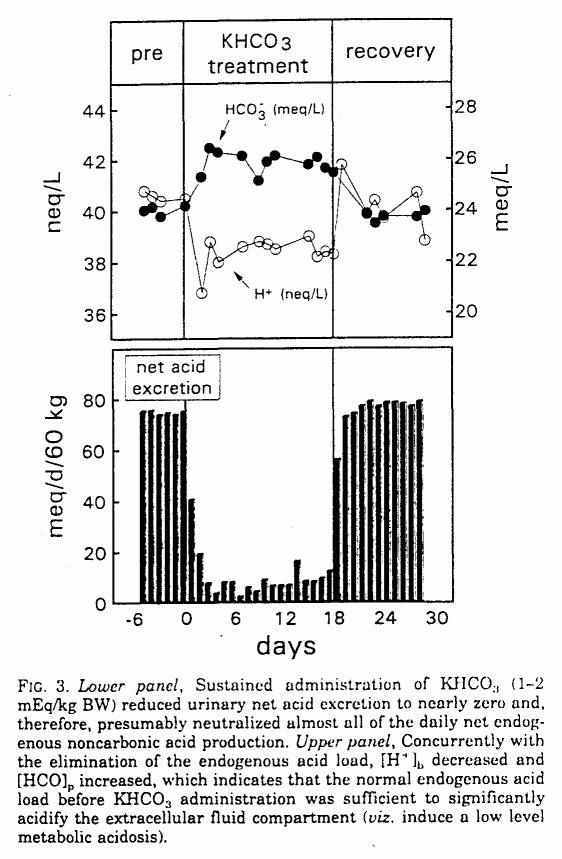
During KHCO3 administration,
[H+]b decreased and [HCO]p
increased significantly, from 40.2 ± 2.2 to 38.1 ±
1.7 mEq/L (P < 0.001) and from 24.2 ± 1.5 to
25.9 ± 1.5 mEq/L (P <0.001), respectively. Net
acid excretion decreased from 75.0 ± 9.7 to 10.2 ±
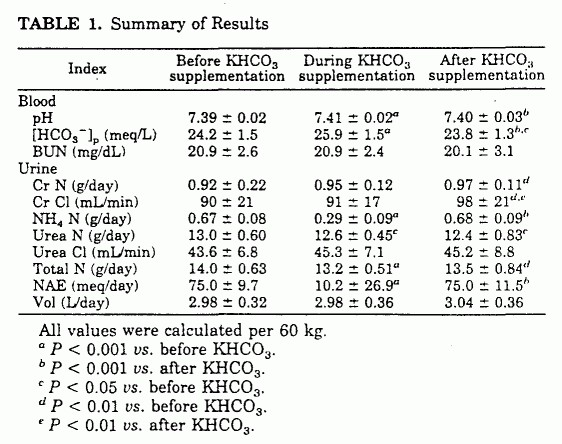
26.9 mEq/day (P < 0.001; Table 1 and Fig. 3). Urinary
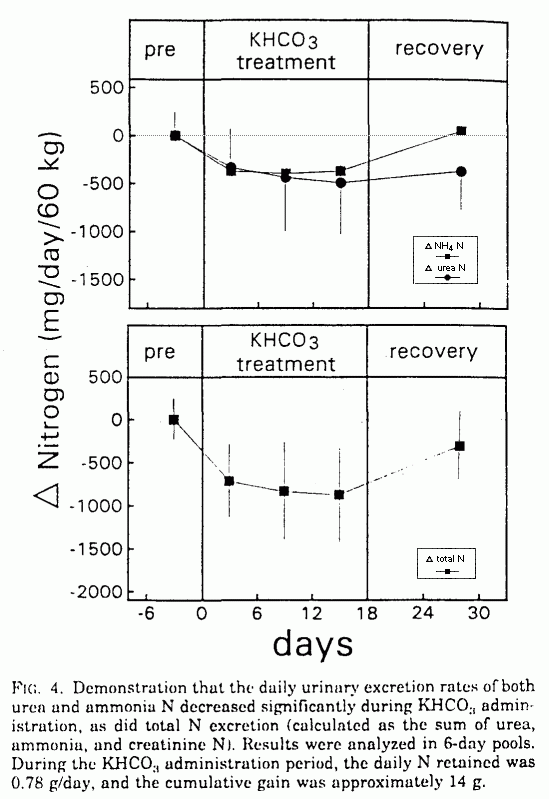
ammonium N (NH4 N), urinary urea N (UUN), and total N
excretion decreased significantly: NH4 N, (671
± 83 to 287 ± 90 mg/day (P < 0.001);
UUN, 12,967 ± 604 to 12,551 ± 451 mg/day
(P < 0.05); and total N, 13,988 ± 625 to
13,193 ± 510 mg/day (P < 0.001; Table 1 and
Fig. 4). Over the 18-day KHCO3 supplementation period,
the cumulative reduction in urinary N excretion was 14.4 g.
Consider that magnitude of cumulative N reduction in terms of
lean body mass (LBM). One kilogram of LBM is equivalent to 32 g N
(25). Therefore, 14 g N in 18 days is equivalent to 0.43 kg LBM
(14 g N ÷ 32 g N/kg LBM) in 18 days. In women, LBM, which
is predominately skeletal muscle, decreases by about 0.22 kg/yr
(20) or about 10.8 g/18 days (0.22 kg/yr X 1000 g/kg X 1 yr/365
days X 18 days). Thus, the KHCO3- cumulative reduction
in N excretion was sufficient to completely offset the expected
age-related cumulative N loss over the same period, with 3.2 g N
to spare. Extrapolating that 3.2 g net gain over 1 yr would
result in a net gain of LBM of 2.0 kg (3.2 g N/18 days x 365
days/yr x 1 kg LBM/32 g N), which would amount to a restoration
of about 1 decade of muscle mass decline. Thus, the magnitude of
the KHCO3-induced N-sparing effect is potentially
sufficient to both prevent continuing loss of muscle mass and
restore previously accrued deficits.
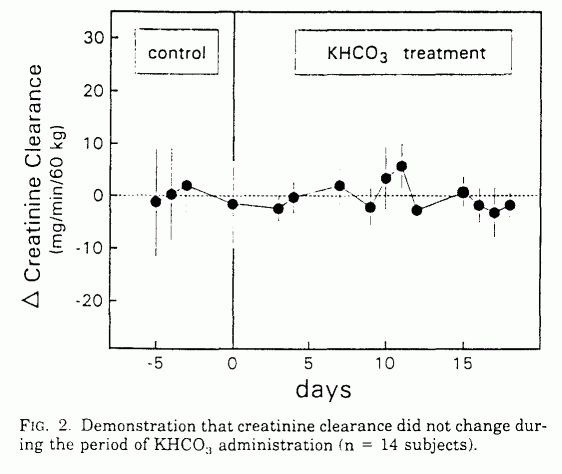
KHCO3 supplementation had no significant effect on
serum creatinine, urinary creatinine, creatinine clearance, serum
urea N, urea clearance, or urine volume (Table 1 and Fig. 2).
Mean body weight was unchanged (+0.2 ± 0.4 kg; P
= NS).
When the KHCO3 supplement was stopped,
[H+]b, [HCO]p, and net acid
excretion promptly returned to baseline (Table 1 and Fig. 3).
Within the period of observation, NH4N excretion
returned to baseline, and UUN and total N excretion started to
increase (Table 1 and Fig. 4).
Discussion
The findings of the present study indicate that in healthy
postmenopausal women: 1) reducing the diet net acid load from
normal to nearly zero with exogenous base (KHCO3)
significantly reduces blood acidity and increases the plasma
bicarbonate concentration, indicating that the unsupplemented
normal diet net acid load was significantly perturbing systemic
acid-base equilibrium, causing a low grade metabolic acidosis; 2)
correction of the diet-dependent metabolic acidosis causes a
significant reduction in urinary N excretion, comprising nearly
equal reductions in urinary ammonia and urea excretion; and 3)
this N-sparing effect is reversed by withholding the exogenous
base.
Because blood acidity decreased significantly and plasma
bicarbonate increased significantly when the diet-dependent net
acid load was eliminated, the preexisting net acid load must have
been significantly perturbing systemic acid-base equilibrium, in
effect causing a low grade metabolic acidosis. Because American
diets typically yield positive nonzero rates of net endogenous
acid production (14), and because the functional integrity of the
kidneys progressively declines during adulthood (24), chronic low
grade age-related metabolic acidosis is the norm in adult humans.
When the net acid load increases from zero to values typical of
American net acid-producing diets, the kidney increases net acid
excretion to a new steady state approaching the new net acid
load, thus stabilizing blood acid-base equilibrium, but not
before a new steady state has developed with significantly
greater blood acidity and lower plasma bicarbonate
concentrations, the levels of which are determined in part by the
magnitude of the acid load and the prevailing functional
integrity of the kidney. Failure to recognize the respective
roles of the diet net acid load and the age-related impaired
renal acid-base regulatory integrity has prevented recognition of
the low grade acidotic state that exists in otherwise healthy
adults, whose acidotic plasma acid-base composition traditionally
has been viewed as normal. Rather, the truly nonacidotic state is
defined as the plasma acid-base composition when the diet net
acid load is zero and renal function is unimpaired, as in young
adulthood.
It is understandably difficult to think “metabolic
acidosis” when the values for plasma acid-base composition
are in the range traditionally considered normal. As the term
metabolic acidosis implies pathophysiological sequelae, if such
sequelae were not present with normal diet net acid loads one
might remain skeptical despite the arguments presented above,
but, in fact, such acidosis-induced pathophysiological conditions
as negative calcium and phosphorus balance and accelerated bone
resorption appear to be a consequence of the normal diet acid
load and are significantly improved by normalizing blood
acid-base composition by neutralizing the diet net acid load with
small amounts of exogenous base (16). As Alpern (26) has noted,
the degree of metabolic acidosis manifested by the plasma
acid-base composition underestimates the severity of the
pathophysiological injury caused by the acid-base disturbance.
With diet-dependent chronic metabolic acidosis, renal and
extrarenal homeostatic adaptations occur that serve to minimize
disturbances in [H+]b and
[HCO]p, but these adaptations themselves are
detrimental. Those include decreased renal citrate production and
excretion (27), hvpercalciuria (28), dissolution of bone (29),
protein catabolism and muscle wasting (1-3), and progression of
renal disease (30). Thus, homeostatic mitigation of the
disturbance of extracellular acid-base equilibrium requires the
body to accept certain deleterious trade-offs (26). Although the
degree of diet-dependent metabolic acidosis is mild as judged by
the degree of perturbation of blood acid-base equilibrium, it
cannot be considered mild as judged by its negative biological
effect.
Chronic metabolic acidosis is a recognized cause of renal N
wasting. N loss occurs both in conditions in which net endogenous
acid production is abnormally high, such as chronic ketoacidosis
induced by dietary carbohydrate deficiency (4, 5), and in
conditions in which the endogenous acid load is not increased but
the acid excretory and/or bicarbonate reabsorptive capacity of
the kidney is impaired, such as the acidosis caused by advanced
renal insufficiency (2, 3, 6, 7). In both conditions, the N
wasting and attendant negative N balance are reversed on
correction of acidosis by the administration of exogenous alkali
(8-11). Consistent with that effect of alkali, in children with
chronic renal acidosis, correction of acidosis with alkali
therapy reverses growth retardation and permits the attainment
and maintenance of normal stature (31).
Metabolic acidosis induces N wasting in part by directly
increasing the rate of protein degradation in skeletal muscle
without commensurately increasing the rate of protein synthesis
(1, 2). That proteolytic effect has been attributed to two
acidosis-induced disturbances in skeletal muscle cells:
stimulation of an ATP- and ubiquitin-dependent proteolytic
pathway (32) and enhancement of the oxidation of proteolytically
released branched chain amino acids (valine, leucine, and
isoleucine), preventing their reuptake for protein synthesis
(33-35). Nonbranched chain amino acids, especially glutamine,
released into the circulation in increased supply, are made
available to the kidney for the generation and excretion of
ammonium, thereby eliminating muscle N and precluding its reuse
for protein synthesis (2, 3, 12, 13).
Additional mechanisms operate during chronic metabolic
acidosis to facilitate renal excretion of ammonium and promote N
wasting. Chronic metabolic acidosis causes an adaptive increase
in renal glutamine extraction and ammonia production (36).
Glutamine becomes the major source of the increased ammonium
excreted in the urine. Acidosis also stimulates hepatic
production of glutamine, which serves to sustain the
acidosis-augmented rates of renal glutamine extraction and
utilization (7, 37-39).
Consistent with the above considerations, in our subjects,
correction of diet-dependent acidosis was accompanied by a
reduction in urinary ammonium excretion, which returned to
control when the acidosis was allowed to recur by discontinuing
the KHCO3 supplement. However, in addition to the
reduction in NH4 N excretion during KHCO3
administration, a sustained reduction in UUN excretion also
occurred, suggesting that the higher pretreatment excretion rates
of urea were contributing to the acidosis-induced N wasting. The
reductions in urea and ammonia excretion contributed about
equally to the N-sparing effect.
Because no detectable reduction occurred in creatinine
clearance with KHCO3 administration, the observed
reduction in urea excretion was not attributable to a reduced
filtered load of urea caused by a reduced glomerular filtration
rate. Similarly, because urine flow was not reduced, reduced urea
excretion was not attributable to increased passive urea
absorption secondary to increased renal water reabsorption.
Otherwise, assuming a urea distribution space equal to total body
water, the resultant near doubling of the serum urea N
concentration would have been easily detected. Alternatively, if
the reduction in urea excretion was due to reduced endogenous net
urea production, the requisite reduction in serum urea N (<0.4
mg / dL) would have been too small to observe. Accordingly, the
reduction in urea excretion during KHCO3
administration is consistent with a reduced net rate of urea
production, but inconsistent with either reduced a glomerular
filtration rate or increased renal urea reabsorption.
We believe that the most straightforward interpretation of the
findings in this study is this. KHCO3 administration
reduced the net endogenous acid production and corrected the
preexisting low grade metabolic acidosis, raising urine pH and
reducing the total rate of renal ammonia production. As a
consequence, both the excretion of ammonia in the urine arid the
delivery of ammonia to the systemic circulation via the renal
vein decreased. The reduction in urinary ammonia contributed
directly to the improvement in N balance. The reduction in
ammonia delivery to the systemic circulation via the renal vein
in addition contributed indirectly to improvement in N balance by
limiting substrate (viz, ammonia) availability for hepatic urea
production (40), thereby reducing external loss of N as urinary
urea. In addition, by correcting the preexisting low grade
metabolic acidosis, KHCO3 decreased the pretreatment
rate of muscle proteolysis, thereby limiting the availability of
amino acids for both urea and ammonia production, further
contributing to the improvement in N balance.
Reduction of urea excretion during alkali administration has
been observed in other acidotic states. Papadoyannakis et
al. (8) reported significant reductions in urinary urea and
total urinary N excretion in response to sodium bicarbonate
administration in patients with renal acidosis. In fasting
subjects with ketoacidosis, administration of bicarbonate salts
reduced urea N excretion significantly in some studies (9), but
not in others (10, 11). Hannaford et al. (9) reported a
significant reduction in urinary urea excretion in response to
the administration of combined sodium bicarbonate and potassium
chloride in fasting obese subjects with ketoacidosis.
Gougeon-Reyburn et al. (10, 11), using a very low
calorie diet (<500 Cal) made up almost exclusively of protein,
found no significant change in urea excretion in response to
either sodium or KHCO3. The reason for the difference
in response of urea excretion between the studies of Hannaford
and Gougeon-Reyburn is not clear.
Whether the renal N-sparing effect of KHCO3
administration translates to improved whole body N balance is not
specifically answered in this study because we did not also
measure fecal N excretion. Fecal N excretion is a small fraction
(<12%) of the total N excretion (25), however, and does not
change with alkali administration (8). Papadoyannakis et
al. (8) found no change in daily measured stool N losses
during sodium bicarbonate treatment of patients with renal
acidosis, although urinary N excretion decreased significantly.
Further studies are necessary to determine the effects of
KHCO3 administration on N balance and LBM.
Acknowledgments
These studies were carried out in the General Clinical
Research Center (GCRC), UCSF (NIH NCRR Grant M0l 00079). This
research was supported in addition by National Institutes of
Health Grants P01DK 39964 and R01DK 32361; University of
California Research Evaluations and Allocation Committee Grant
MSC-22; UCSF Academic Senate Grant; and gifts from Church &
Dwight Co., Inc., and the Emil Mosbacher, Jr., Foundation. We
want to thank the nursing, dietary, and laboratory staff of the
GCRC for their assistance in carrying out these studies.
References
1. Garibotto G, Russo R, Sofia A, et al. 1996 Muscle protein
turnover in chronic renal failure patients with metabolic
acidosis or normal acid-base balance. Miner Electrolyte Metab.
22:58-61.
2. May RC, Kelly RA, Mitch WE. 1986 Metabolic acidosis
stimulates protein degradation in rat muscle by a
glucocorticoid-dependent mechanism. J Clin Invest.
77:614-621.
3. Williams B, Layward E, Walls J. 1991 Skeletal muscle
degradation and nitrogen wasting in rats with chronic metabolic
acidosis. Clin Sci. 80:457-462.
4. Vazquez JA, Adibi SA. 1992 Protein sparing during treatment
of obesity: ketogenic versus nonketogenic very low calorie diet.
Metabolism. 41:406-414.
5. Bell JD, Margen S, Calloway DH. 1969 Ketosis, weight loss,
uric acid, and nitrogen balance in obese women fed single
nutrients at low caloric levels. Metabolism. 18:193-208.
6. May RC, Kelly RA, Mitch WE. 1987 Mechanisms for defects in
muscle protein metabolism in rats with chronic uremia. Influence
of metabolic acidosis. J Clin Invest. 79:1099-1103.
7. Welbourne TC, Joshi S. 1994 Enteral glutamine spares
endogenous glutamine in chronic acidosis. J Parenter Enter Nutr.
18:243-247.
8. Papadoyannakis NJ, Stefanidis CJ, McGeown M. 1984 The
effect of the correction of metabolic acidosis on nitrogen and
potassium balance of patients with chronic renal failure. Am J
Clin Nutr. 40:423-427.
9. Hannaford MC, Leiter LA, Josse RG, Goldstein MB, Marliss
EB, Halperin ML. 1982 Protein wasting due to acidosis of
prolonged fasting. Am J Physiol. 243: E251-E256.
10. Gougeon-Reyburn R, Laraviere F, Marliss EB. 1991 Effects
of bicarbonate supplementation on urinary mineral excretion
during very low energy diets. Am J Med Sci. 302:67-74.
11. Gougeon-Reyburn R, Marliss EB. 1989 Effects of sodium
bicarbonate on nitrogen metabolism and ketone bodies during very
low energy protein diets in obese subjects. Metabolism.
38:1222-1230.
12. Guder WG, Haussinger D, Gerok W. 1987 Renal and hepatic
nitrogen metabolism in systemic acid base regulation. J Clin Chem
Biochem. 25:457-466.
13. Cersosimo E, Williams PE, Radosevich PM, Hoxworth B, Lacy
WW, Abumrad NN. 1986 Role of glutamine in adaptations in nitrogen
metabolism during fasting. Am J Physiol. 250:E622-E628.
14. Kurtz I, Maher T, Hulter HN, Schambelan M, Sebastian A.
1983 Effect diet on plasma acid-base composition in normal
humans. Kidney Int. 24:670-680.
15. Lennon EJ, Lemann Jr J, Litzow JR. 1966 The effect of diet
and stool composition on the net external acid balance of normal
subjects. J Clin Invest. 45:1601-1607.
16. Sebastian A, Harris ST, Ottaway JH, Todd KM, Morris Jr RC.
1994 Improved mineral balance and skeletal metabolism in
postmenopausal women treated with potassium bicarbonate. N EngI J
Med. 330:1776-1781.
17. Licata AA, Bou E, Bartter FC, Cox J. 1979 Effects of
dietary protein on urinary calcium in normal subjects and in
patients with nephrolithiasis. Metabolism. 28:895-900.
18. Breslau NA, Brinkley L, Hill KD, Pak CYC. 1988
Relationship of animal protein-rich diet to kidney stone
formation and calcium metabolism. J Clin Endocrinol Metab.
66:140-146.
19. Abelow BJ, Holford TR, Insogna KL. 1992 Cross-cultural
association between dietary animal protein and hip fracture: a
hypothesis. Calcif Tissue Int. 50:14 -38.
20. Forbes GB. 1976 The adult decline in lean body mass. Hum
Biol. 48:161-173.
21. Novak LP. 1972 Aging, total body potassium, fat-free mass,
and cell mass in males and females between ages 18 and 85 years.
J Gerontol. 27:438-443.
22. Frassetto L, Sebastian A. 1996 Age and systemic acid-base
equilibrium: analysis of published data. J Gerontol.
51A:B91-B99.
23. Frassetto L, Morris Jr RC, Sebastian A. 1996 Effect of age
on blood acid-base composition in adult humans: role of
age-related renal functional decline. Am J Physiol.
271:1114-1122.
24. Davies DF, Shock NW. 1950 Age changes in glomerular
filtration rate, effective renal plasma flow, and tubular
excretory capacity in adult males. J C. Invest. 29:496-507,
25. Anonymous. 1988 In: Shils ME, Young VR, eds. Modern
nutrition in health and disease, 7th ed. Philadelphia: Lea and
Febiger; 1-1694.
26. Alpern RJ. 1995 Trade-offs in the adaptation to acidosis.
Kidney Int. 47:1205-1225.
27. Gordon EE. 1963 Effect of acute metabolic acidosis and
alkalosis on acetate and citrate metabolism in the rat. J Clin
Invest. 42:137-142.
28. Lemann Jr J, Litzow JR, Lennon EJ. 1966 The effects of
chronic acid loads in normal man: further evidence for
participation of bone mineral in the defense against chronic
metabolic acidosis. J Clin Invest. 45:1608-1614.
29. Barzel US, Jowsey J. 1969 The effects of chronic acid and
alkali administration on bone turnover in adult rats. Clin Sci.
36:517-524.
30. Nath KA, Salahudeen AK, Clark EC, Hostetter MK, Hostetter
TH. 1992 Role of cellular metabolites in progressive renal
injury. Kidney Int. 38(Suppl):S109- S113.
31. McSherry E, Morris Jr RC. 1978 Attainment and maintenance
of normal stature with alkali therapy in infants and children
with classic renal tubular acidosis. J Clin Invest.
61:509-527.
32. Mitch WE, Medina R, Grieber S, et al. 1994 Metabolic
acidosis stimulates muscle protein degradation by activating the
adenosine triphosphate-dependent pathway involving ubiquitin and
proteasomes. J Clin Invest. 93:2127-2133.
33. May RC, Masud T, Logue B, Bailey J, England BK. 1992
Metabolic acidosis accelerates whole body protein degradation and
leucine oxidation by a glucocorticoid-dependent mechanism. Miner
Electrolyte Metab. 18:245-249.
34. May RC, Masud T, Logue B, Bailey J, England B. 1992
Chronic metabolic acidosis accelerates whole body proteolysis and
oxidation in awake rats. Kidney Int. 41:1535-1542.
35. May RC, Hara Y, Kelly RA, Block KP, Buse MG, Mitch WE.
1987 Branched-chain amino acid metabolism in rat muscle: abnormal
regulation in acidosis. Am J Physiol. 252:E712-E718.
36. Pitts RF. 1973 Production and excretion of ammonia in
relation to acid-base balance. In: Orloff J, Berliner RW, eds.
Handbook of physiology, sect 8. Washington DC: American
Physiological Society; 455-496.
37. Haussinger D. 1990 Organization of hepatic nitrogen
metabolism and its relation to acid-base homeostasis. Klin
Wochenschr. 68:1096-1101.
38. Oliver J, Koelz AM, Costello J, Bourke A. 1977 Acid-base
induced alterations in glutamine metabolism and ureogenesis in
perfused muscle and liver of the rat. Eur J Clin Invest.
7:445-449.
39. Almond MK. Iles RA, Cohen RD. 1992 Hepatic glutamine
metabolism and acid-base regulation. Miner Electrolyte Metab.
18:237-240.
40. Cheema-Dhadli S, Jungas RL, Halperin ML. 1987 Regulation
of urea synthesis by acid-base balance in vivo: role of
NH3 concentration. Am J Physiol. 252: F221-F225.
Received May 10, 1996. Revision received September 13, 1996.
Accepted September 20, 1996.
Address all correspondence and requests for reprints to:
Anthony Sebastian, M.D., Box 0126, University of California, San
Francisco, California 94143.
This page was first uploaded to The Magnesium Web Site on
December 21, 2002
http://www.mgwater.com/
