The Lancet, Vol. 340; July 18, 1992
REVIEW ARTICLE
Protein processing in lysosomes: the new therapeutic target
in neurodegenerative disease
R. JOHN MAYER, MICHAEL LANDON, LAJOS LASZLO, GRAHAM LENNOX,
JAMES LOWE
A little recognised feature of neurons is their large
complement of lysosomes. Studies of the accumulation of the
abnormal isoform of the prion protein (PrPSC) in the
prion encephalopathies and the formation of β/A4 protein
from its precursor in Alzheimer’s disease suggest that
generation of these key proteins takes place in lysosome-related
organelles. The release of hydrolytic enzymes from lysosomes may
be a primary cause of neuronal damage.
Although molecular genetic approaches have identified protein
mutations central to the main neurodegenerative disease, cell
biological observations are now beginning to unravel the
intracellular pathways involved in the molecular pathogenesis of
neurodegeneration: as a result, it is now appropriate to consider
therapeutic manipulation of the lysosomal system as an approach
to treatment.
Introduction
Neurons do not replicate in adult life, so they need an
efficient way of turning over proteins and dealing with any
abnormal proteins. To this end they possess a very well-developed
lysosome system. Evidence is accumulating to suggest that
abortive attempts to degrade proteins within this system lie at
the centre of the pathogenesis of some of the major
neurodegenerative diseases of man. These include Alzheimer
disease and the prion encephalopathies such as Creutzfeldt-Jakob
disease where abnormal amyloid (β/A4) and prion
(PrPSC)proteins, respectively, are deposited in and
around neurons. This in turn opens up the possibility of new
therapeutic strategies, aimed at altering lysosomal protein
processing.
Lysosome system
Lysosomes are the most familiar part of the large system of
acid-containing vesicles that enable cells to digest unwanted
material. They are characterised by specific hydrolases (eg,
(β-glucuronidase) which are most active at low pH. Other
components of this acidic vesicle system include endosomes
(vesicles formed after membrane internalisation during
receptor-mediated endocytosis), multivesicular and
tubulovesicular bodies (which may form by the surface
invagination of endosomes), autophagic vacuoles (formed within
cells to isolate unwanted organelles), and nascent
hydrolase-containing vesicles derived from the protein-packaging
Golgi apparatus.1
Recent evidence suggests that the lysosome system interacts
closely with cell stress proteins. Cell stress
proteins—also known as heat-shock proteins (HSP) after one
form of cell stress used in early experiments—are highly
conserved and have roles in normal cell activity as well as in
the protective response to cell damage. They include ubiquitin, a
central co-factor in protein degradation, and HSP 70,2
which acts as a molecular “chaperone”, facilitating
the folding and transport of proteins across different
compartments within the cell.3 Initially thought of as
cytosolic proteins, both are also found within lysosome related
organelles. Immunogold electronmicroscopy has shown that normal
lysosomes contain both free ubiquitin4 and
ubiquitin-protein conjugates5-7 and that these
conjugates accumulate excessively in lysosomes whose function has
been compromised by drugs.8 The precise function of
ubiquitin and HSP 70 in lysosomes is not clear, although it
presumably relates to the regulation of protein degradation.
Certainly cells with a mutation of the ubiquitin activating
enzyme El can no longer degrade proteins in
lysosomes.9 In addition, ubiquitin and HSP 70 are
useful markers of the lysosome system in both health and
disease.
Ubiquitin-protein conjugates in health and
disease
Deposits of ubiquitin-protein conjugates are seen within the
neuropil of the normal elderly human brain in numbers that
increase with age.10,11 These are nerve cell processes
(neurites) packed with ubiquitin-immnunoreactive lysosome-related
dense bodies (fig 1). Similar lysosome-related accumulations of
ubiquitinated proteins are found in several pathological
conditions. Dot-like structures are seen in the neuropil in mouse
scrapie (an animal model of prion encephalopathy), together with
coarser structures adjacent to neurons which resemble autophagic
vacuoles.12 The dot-like structures develop very early
after infection, at the time when abnormal prion protein first
becomes detectable.13 Later on larger deposits of
ubiquitinated proteins develop in structures resembling
lysosomes, and in dystrophic neurites around amyloid plaques (fig
2). Identical structures are seen in the equivalent human prion
diseases (Creutzfeldt-Jakob disease14 and
Gerstmann-Straussler-Scheinker syndrome15).
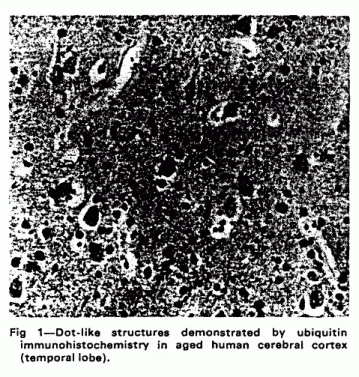
Similarly in Alzheimer disease deposits of ubiquitin-protein
conjugates are seen within dystrophic neurites around senile
plaques; these appear to occur in membranous and vesicular dense
bodies resembling lysosomes.16 Other characteristic
elements of the pathology of Alzheimer disease also contain
ubiquitinated proteins, such as the neurofibrillary
tangles17-19 and areas of granulovacuolar degeneration
(the latter again bearing some topographical resemblance to
lysosomal structures20).
It has generally been assumed that these observations are
epiphenomena, the persistence of ubiquinated proteins and their
accumulation into lysosomal structures representing an
appropriate and cytoprotective (if ultimately ineffective)
response to the disease process. There is, however, now evidence
to suggest that this response may itself be pathogenic and play a
central part in causing neuronal damage.
Lysosome-related organelles, prion
encephalopathies, and Alzheimer disease
Recent advances in identifying the prion protein
(PrPC) and understanding its molecular
genetics21,22 have shed little light upon the
mechanisms which underlie its transformation into an abnormal
isoform, its accumulation, and the subsequent development of
spongiform pathology. The normal prion protein is a membrane
protein found in a variety of cell types, including
neurons.23 Cell culture studies of scrapie have shown
that the abnormal isoform of prion protein appears within
intracellular organelles24,25 which include
lysosome-related structures.26 These are capable of
partly truncating the protein27 and may provide an
environment in which the abnormal isoform is generated from the
normal prion protein. A hypothetical scheme for this is shown in
fig 3.
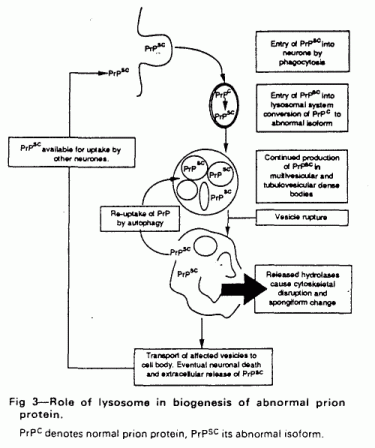
During natural or experimental infection with prion disease
the abnormal prion protein will be taken up into the
lysosome-related system by some phagocytic process. This will
inevitably in portions of cell membrane (including normal prion
protein) into endosomes; both normal and abnormal prion isoforms
will then enter multivesicular bodies. In the acidic denaturing
milieu, unfolded and part-fragmented prion protein molecules will
be able to undergo the secondary structural interaction which is
thought to result in the generation of further abnormal prion
protein. Eventually a critical level of abnormal prion protein
will accumulate, first disrupting the lysosomal membrane and then
releasing hydrolases into the cell. Many of these enzymes retain
activity at neutral pH and will cause neuronal damage.
Ultimately, when most lysosomes in a neuron are overwhelmed by
accumulated abnormal prion protein, the neuron will die and
release abnormal prion protein to be taken up by other neurons
through phagocytosis. This would lead to an exponential increase
of abnormal prion protein culminating in neurodegeneration. In
this context the lysosomal system is acting as the
“bioreactor” for the formation of abnormal prion
protein.
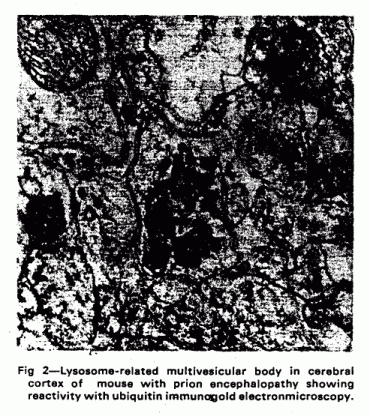
This scheme is supported by pathological studies of murine
scrapie, where immunogold electronmicroscopy has shown an
accumulation of lysosome-related multivesicular, tubulovesicular
and dense bodies containing hydrolases such as
β-glucuronidase as well as HSP 70, ubiquitin conjugates, and
abnormal priori protein. All of these can be seen spilling out of
the lysosome-related vesicles into areas of rarefaction which are
thought to be the precursors of the larger areas of spongiform
change which characterise the prion
encephalopathies.28 Similar ubiquitin deposits and
lysosomes can be seen in immunohistochemically adjacent to
spongiform lesions in Creutzfeldt disease.29
A similar model can be constructed for Alzheimer disease. Here
the amyloid precursor protein is thought to have a central
role.30 It is concentrated in neuronal lysosomes in
both normal and Alzheimer disease brain,31,32 where it
is subject to partial degradation.33,36 Again the
acidic denaturing interior of lysosome-related structures
provides an ideal environment for the transformation of the
amyloid precursor protein into smaller β/A fragments and for
the secondary structural interactions which are required for
amyloid formation. In Alzheimer disease the fate of the
bioreactor lysosomes may be slightly different; fusion of the
lysosomes with the neuronal membrane would lead to the expulsion
of their amyloid contents before neuronal death and the
accumulation of extracellular amyloid, characteristically seen at
the centre of senile plaques. An ejection mechanism of this kind
is well-documented in other contexts—for example, in
exporting transferrin receptors from maturing
reticulocytes.34
New therapeutic targets?
The nervous system, with its long-lived neurons, is vitally
dependent on an effective lysosomal waste disposal system. Unlike
other cell types, neurons cannot divide to replace cells that
have died through the accumulation of indigestible material.
Processing of proteins may become an increasing burden with
ageing, and this accounts for the development of lysosome-related
ubiquitinated dot-like structures in elderly neurons. These
probably represent a common neuronal response to ageing and cell
injury, reflecting activation of the lysosome-related system.
Protein processing in the lysosome-related system, modulated
by the cell stress proteins, may be crucial in disease states.
Modification of normal neuronal precursor proteins and the
accumulation or deposition of their abnormal products appears to
be a central part of neurodegenerative diseases like Alzheimer
disease and the prion encephalopathies. The lysosome system
provides the only intracellular environment capable of performing
this pathological processing, and the recent observations
reviewed here suggest that it lies at the heart of the
pathogenesis of these diseases. This raises the possibility that
interference with protein processing in lysosome related
organelles might confer therapeutic benefit. In Alzheimer disease
it might be possible to block the conversion of amyloid precursor
protein to the β/A4 fragments and amyloid, so preventing
deposition of the latter within and around neurons. In the priori
encephalopathies it might be possible to prevent the rupture of
lysosome-related bodies into neurons and halt the development of
spongiform change. In both cases such interventions might
usefully slow the progression of the disease. Drugs that
influence lysosomal function already exist, as do transgenic
models for at least the prion encephalopathies,35
which would allow this strategy to be rapidly put to the test. In
the absence of any other effective treatment, we suggest that
approaches to lysosomal intervention merit serious
consideration.
We thank the Wellcome Trust, Parkinson’s Disease
Society, and Motor Neurone Disease Association for support of
some of this work.
REFERENCES
1.Holzmann E. Lysosomes. New York: Plenum Press, 1989.
2. Chiang H-L, Terlecky S, Plant C, et al. A role for a 70
kilodalton heat shock protein in lysosomal degradation of
intracellular proteins. Science 1989;
246: 282-84.
3. Lowe J, Mayer RJ. Ubiquitin, cell stress and diseases of
the nervous system. Neuropothol Appl Neurobiol 1990;
16: 281-91.
4. Schwartz AL, Ciechanover A, Brandt RA, et al.
Immunoelectron microscopic localization of ubiquitin in hepatoma
cells. EMBOJ 1988; 7: 2961-66.
5. Laszlo L, Doherty FJ, Osborn NU, et al. Ubiquitinated
protein conjugates are specifically enriched in the lysosomal
system of fibroblasts. FEBS Lett 1990;
261: 365-68.
6. Laszlo L, Tuckwell J, Self T, et al. The latent membrane
protein-1 in Epstein-Barr virus-transformed lymphoblastoid cells
is found with ubiquitin-protein conjugates and heat-shock protein
70 in lysosomes oriented around the microtubule organising
centre. J Pathol 1991; 164: 203-14.
7. Laszlo L, Doherty FJ, Watson A, et al. Immunogold
localisation of ubiquitin-protein conjugates in primary
(azurophilic) granules of polymorphonuclear neutrophils. FEBS
Lett 1991,279: 175-78.
8. Doherty FJ, Osborn NU, Wassall JA, et al. Ubiquitin-protein
conjugates accumulate in the lysosomal system of fibroblasts
treated with cysteine proteinase inhibitors. Biochem J
1989; 263: 47-55.
9. Gropper R, Brandt RA, Elias S, et al. The
ubiquitin-activating enzyme, El, is required for stress-induced
lysosomal degradation of cellular proteins.J Biol Chem
1991; 266: 3602-10.
10. Migheli A, Attanasio A, Pezzulo T, et al. Age-related
ubiquitin deposits in dystrophic neurites: an immunoelectron
microscopic study. Neuropathol Appl Neurobiol 1992;
121:55-58.
11. Dickson DW, Wertkin A, Kress Y, et al. Ubiquitin
immnunoreactive structures in normal human brains. Lab
Invest 1990; 63: 87-99.
12. Lowe J, McDermott H, Kenward N, et al. Ubiquitin conjugate
immunoreactivity in the brains of scrapie infected mice. J
Pathol 1990; 162: 61-66.
13. Lowe J, Fergusson J, Kenward N, et al. Immunoreactivity to
ubiquitin-protein conjugates is present early in the disease
process in the brains of scrapie infected mice.J Pathol
(in press).
14. Suenaga T, Hirano A, Llena JF,et al. Ubiquitin
immunoreactivity in kuru plaques in Creutzfeld-Jakob disease.
Ann Neural 1990; 28: 174-77.
15. Migheli A, Attanasio A, Vigliani MC, et al. Dystrophic
neurites around amyloid plaques of human patients with
Gerstmann-Straussler-Scheinker disease contain ubiquitinated
inclusions. Neurosci Lett 1991; 121:
55-58.
16. Dickson DW, Wertkin A, Mattiace LA, et al. Ubiquitin
immunoelectron microscopy of dystrophic neurites in cerebellar
senile plaques of Alzheimer's disease. Acta Neuropathol
1990; 79: 486-93.
17. Mori H, Kondo J, Ihara Y. Ubiquitin is a component of
paired helical filaments in Alzheimer’s disease.
Science 1987; 235: 1641-44.
18. Perry G, Friedman R, Shaw G, et al. Ubiquitin is detected
in neurofibrillary tangles and senile plaque neurites of
Alzheimer disease brains. Proc Natl Acad Sci USA 1987;
84: 3033-36.
19. Cole GM, Timiras PS. Ubiquitin-protein conjugates in
Alzheimer’s lesions. Neurosci Len 1987;
79: 207-12.
20. Lowe J, Blanchard A, Morrell K, et al. Ubiquitin is a
common factor in intermediate filament inclusion bodies of
diverse type in man,including those of Parkinson’s disease,
Pick’s disease, and Alzheimer’s disease, as well as
Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies
in muscle, and Mallory bodies in alcoholic liver disease.J
Pathol 1988; 155: 9-15.
21. Prusiner SB. Molecular biology of prion diseases.
Science 1991; 252:1515-22.
22. Brown P, Goldfarb LG, Gajdusek DC. The new biology of
spongiform encephalopathy: infectious amyloidoses with a genetic
twist. Lancet 1991; 337: 1019-22.
23. Stahl N, Borchelt DR, Hsiao KK, et al. Glycolipid
modification of the scrapie prion protein. Cell 1987;
51: 229-40.
24. Borchelt DR, Scott M, Taraboulos A, et al. Scrapie and
cellular prion proteins differ in their kinetics of synthesis and
topology in cultured cells.J Cell Biol 1990;
110:743-52.
25. Taraboulos A, Serban D, Prusiner SB. Scrapie prion
proteins accumulate in the cytoplasm of persistently infected
cultured cells.J Cell Biol 1990;
110:2117-32.
26. McKinley MP, Taraboulos A, Kenaga L, et al.
Ultrastructural localisation of scrapie prions in cytoplasmic
vesicles of infected cultured cells. Lab Invest 1991;
65: 622-30.
27. Caughey B, Raymond GJ, Ernst D, et al. N-terminal
truncation of the scrapie-associated form of PrP by lysosomal
protease(s): implications regarding the site of conversion of PrP
to the protease-resistate state. J Virol 1991;
65: 6597-603.
28. Laszlo L, Lowe J, Self T, et al. Lysosomes are key
organelles in the pathogenesis of prion encephalopathies.J
Pathol 1992; 166: 333-41.
29. Ironside JW, Bell JE, McCardle L, et al. Neuronal and
glial reactions in Creutzfeldt-Jakob disease. Neuropathol
Appl Neurobiol 1992; 18: 295.
30. Wright A, Goedert M, Hastie N. Beta amyloid resurrected.
Nature 1991; 349: 653-54.
31. Benowitz LI, Rodriguez W, Paskevich P, et al. The amyloid
precursor protein is concentrated in neuronal lysosomes in normal
and Alzheimer disease subjects. Exp Neural 1989;
106: 237-50.
32. Kawai M, Cras P, Richey P, et al. Subcellular localisation
of amyloid precursor protein in senile plaques of
Alzheimer’s disease. Am J Pathol 1992;
140: 947-58.
33. Golde TE, Estus S, Younkin LH, et al. Processing of the
amyloid precursor protein to potentially amyloidogenic
derivatives. Science 1992; 255:
728-30.
34. David JQ, Dansereau D, Johnstone RM, et al. Selective
externalization of an ATP-binding protein structurally related to
clathrin uncoating ATPase heat shock protein in vesicles
containing terminal transferrin receptors during reticulocyte
maturation. J Biol Chem 1986; 261:
15368-71.
35. Hsaio K, Scott M, Foster D, et al. Spontaneous
neurodegeneration in transgenic mice with mutant prion protein.
Science 1990; 250: 1587-60.
36. Haas C, Koo EH, Mellon A, et al Targeting of cell-surface
β-amyloid precursor protein to lysosomes alternative
processing into amyloid bearing fragments. Nature 1992;
357: 500-03.
ADDRESSES: Departments of Biochemistry (Prof R. J. Mayer, DSc,
M. Landon. PhD, L. Laszlo, PhD). Pathology (J. Lowe, MRCPath),
and Neurology (G. Lennox, BM), University of Nottingham Medical
School. Queen’s Medical Centre, Nottingham NG7 2UH, UK.
Correspondence to Prof R. John Mayer.
This page was first uploaded to The Magnesium Web Site on
December 18, 2002
http://www.mgwater.com/
