MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part III: Chapter 13
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
13
Renal Damage Caused by Magnesium Deficiency
Metastic calcification, frequently involving the kidneys, is not infrequent in patients with hypercalcemia, whether of dietary or metabolic derivation, because of osteolytic processes, or as a result of therapy. The study by B. S. W. Smith and Nisbet (1968), which showed that magnesium-deficient rats develop nephrocalcinosis, and later osteoporosis, is an appropriate reference for the transition from bone damage to renal damage of magnesium deficiency.
13.1. Experimental Magnesium Deficiency
The diets contrived to be magnesium deficient are almost always imbalanced in other constituents as well. The early diets were usually rich in fats, calcium, phosphorus, and vitamin D, which were effective in producing acute signs of magnesium depletion rapidly (Kruse et al., 1932) and also produced severe renal glomerular and tubular damage that was most extensive at the junction of the cortex and medulla (Cramer, 1932; Brookfield, 1934). Modifications of that diet (designed specifically to produce hypercholesterolemia and atherosclerosis) also produced renal damage (Hellerstein et al., 1957; Gottlieb et al., 1959; Vitale et al., 1959). There was deposition of calcium microliths in the lumina of the collecting tubules that was accompanied by tubular dilatation, and flattened epithelium. High dietary magnesium (96mg Mg/100 g of diet) abolished the renal tubular calcification, regardless of the amount of calcium fed, in the animals not loaded with cholesterol and cholic acid, and decreased it in fat-loaded rats.
With less imbalanced diets, designed to produce subacute magnesium deficiency (Watchorn and McCance, 1937), rats developed occasional to more frequent calcareous deposits scattered throughout the renal cortex and medulla. Those with most severe damage had extensive calcareous casts and obliteration of the epithelium of the straight and collecting tubules, but no glomerular changes. Greenberg et al. (1938), also using a less imbalanced diet that did not produce signs of acute deficiency and that contained neither excess phosphate nor very high doses of vitamin D, but was high in calcium (Tufts and Greenberg, 1937), found that prolonged magnesium deprivation of rats produced corticomedullary necrosis and calcinosis involving both the tubular cells and lumina. They attributed the renal calcinosis to the high calcium/magnesium ratio. Greenberg (1939) later attributed part of the severe manifestations of the magnesium-deficiency syndrome (including the renal calcinosis) in the early studies to the inadequacy of vitamins B2 and B6 in the vitamin-B-complex supplements then available. The concomitant magnesium and pyridoxine deficiencies might be relevant to calcium oxalate deposition in the kidneys, magnesium being a cofactor in vitamin B6 metabolism (Review: Durlach, 1969b), oxalate excretion increasing in vitamin B6 deficiency (Gershoff et al., 1959), and a combination of high magnesium and vitamin B6 being useful in decreasing calcium oxalate and apatite nephrocalcinosis and urolithiasis (Gershoff and Andrus, 1961; Gershoff and Prien, 1967). Gershoff and Andrus (1961) also showed that the amount of magnesium usually provided control rats (400 ppm) did not completely prevent formation of apatite salts in the kidneys. Tenfold higher intakes were completely protective.
Most of the magnesium-deficiency data derived from rat studies have been obtained with diets rich in calcium and phosphorus, although the marked imbalances in dietary Ca/Mg and P/Mg are rarely noted. Usually they provided from 600/1 to 60/1 ratios of Ca/Mg. For example, rats reported by Hess et al. (1959) were fed a diet delivering 18 mmol Mg/kg of diet and 150 mmol Ca; the deficient group were given 0.25 mmol Mg. They had mitochondrial swelling of tubular cells (observed as early as 3 days of magnesium deprivation) in the distal segment of the convoluted tubule and extending to the thick descending limb. By 6 days, Henle's loop was also involved. Tubular necrosis was noted by 12 to 20 days, and there were calcium deposits intracellularly and in the lumina, forming calcareous casts. The semisynthetic magnesium-deficient diet provided by Mishra (l960a,b) provided a similar Ca/Mg ratio, and caused decreased renal mitochondrial count and increased tubular calcinosis. With an approximately tenfold less disparity between dietary calcium and magnesium, tubular lesions developed in the renal cortex and at the corticomedullary junction by the day 8 of magnesium deficiency (Kashiwa, 1961). Some of the tubular cells were hypertrophied and had vacuolated cytoplasm, others were flattened, and there were numerous calcareous deposits, especially at the corticomedullary junction. Comparable changes, with clumping of renal tubular mitochondria, were correlated with functional renal defects after as little as a week of magnesium depletion (W. O. Smith et al., 1962). The rats exhibited a decreased ability to concentrate and acidify urine and a marked phosphaturia.
Sauberlich and Baumann (1949) found that mice fed diets deficient in thiamine, pyridoxine, or magnesium had aminoaciduria. In a study of chicks and rats (with a Ca/Mg ratio, even in the magnesium-deficient group of rats that was less imbalanced, about 40/1; Bunce et al. (1963) showed that sixfold higher intakes of magnesium were necessary to prevent nephrocalcinosis and aminoaciduria that were seen in the deficient groups. Progressively increased aminoaciduria was also produced in rats on the usual high Ca/Mg dietary ratios of magnesium deficiency studies as the depletion developed (Mazzocco et al., 1966).
Noted in most of the cited magnesium-deficiency studies were the intraluminal calcareous deposits in the corticomedullary area, and the damage to the tubular epithelium. The characteristic early lesion has been described as microliths in the thin limb, the bend of the loop, and the ascending limb of the loop of Henle (ALLH) (Whang et al., 1962; Welt, 1964; Oliver et al., 1966; Schneeberger and Morrison, 1965, 1967; Whang et al., 1969). Ko et al. (t962) reported that rats on a magnesium-deficient diet that provided twice as much calcium as phosphorus developed the typical intraluminal and cellular deposits of calcium phosphate, but that the ALLH was not involved unless there was phosphate loading, as well. Schneeberger and Morrison (1967) showed that the ALLH lesions of magnesium deficiency were intensified by phosphate loads. Similar intraluminal lesions have also been seen in the bend of the loop and in the ALLH of early experimental hyperparathyroidism (Epstein, 1960) and vitamin D toxicity (Epstein et al., 1958; Kent et al., 1958; Veltman, 1959; Potvliege, 1962). This observation is not surprising since both hyperparathyroidism and hypervitaminosis D increase blood and thus urinary loads of calcium, and cause magnesium loss.
Damage to the ALLH by primary or secondary magnesium deficiency creates a situation that intensifies the magnesium deficit. Micropuncture studies have shown that most active renal tubular reabsorption of magnesium occurs at this site (Wen et al., 1970, 1971; Brunette et al., 1974, 1975; Dirks and Quamme, 1978; Quamme et al., 1976/1980). Thus, damage to the cells of the ALLH can cause renal tubular magnesium wasting. The clinical significance of treatment of hypomagnesemic hypocalcemia with calcemic agents or phosphates is discussed elsewhere in this volume.
13.2. Intensification of Magnesium Deficiency and Renal Damage by Excess Vitamin D (Animal)
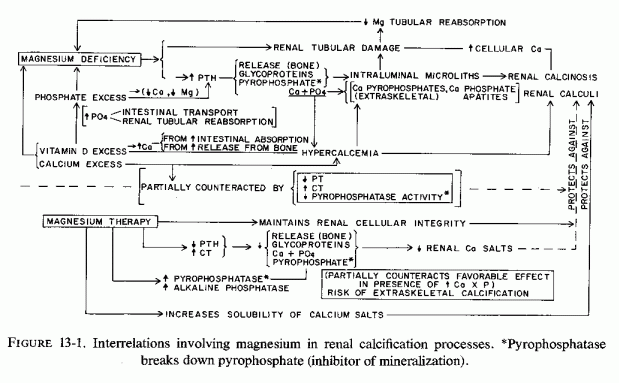
Vitamin D toxicity, with or without high calcium intakes, has long been known to cause soft tissue damage. The cardiovascular lesions have attracted most notice. Even brain damage and calcification have been described, both in test animals and in infantile hypercalcemia (Review: Seelig, 1969b). How much of the total renal damage of most experimental magnesium-deficiency studies is caused by relative or absolute vitamin D excess, and how much might be due to excess phosphate intake or tubular reabsorption, each of which intensifies magnesium loss and increases mobilization of bone constituents has not been resolved (Fig. 13-1). The answer must await definitive studies that evaluate the effects of each agent, with the others kept at the amounts necessary to avoid inducing specific deficiencies or imbalances. Such might evoke hormonal responses that could obfuscate the effect of the mineral under investigation.
Konetzki et al. (1962) showed sequential accumulation of calcium and mucopolysaccharides in nephrocalcinosis due to vitamin D toxicity. The renal deposition of calcium started before the kidneys began to accumulate radioactively tagged sulfur. After the process had started the 35S uptake intensified. (Giacomelli et al. (1964) observed that calcium deposited as hydroxyapatite crystals intraluminally in the proximal convoluted renal tubules and in the cytoplasmic vacuoles of the tubular cells of rats poisoned by vitamin D. They consider the crystallization process to be induced by deposition of mucopolysaccharides (derived, like the calcium and phosphate, from the bone, dissolution of which is caused by hypervitaminosis D). These changes are very much like those described in magnesium-deficient rats: tubular calcium phosphate on a glycoprotein matrix (Bunce and Bloomer, 1972). As with that of magnesium deficiency, the initial lesion of vitamin D nephrotoxicity is proposed to be cytochemical and cytologic alterations (Scarpelli, 1966). The earliest consistent changes, by light microscopy, was increased atypical cytoplasmic vacuoles in the proximal tubular cells [manifest within 24 hours after a single massive oral dose (45,000 units) of vitamin D]. Slight mitochondria damage was also seen. Intracellular edema and marked cellular distortion developed after four doses. Calcific deposits were first seen after six doses of vitamin D, and involved the tubules of the corticomedullary junction. At this time there was marked mitochondrial damage. There was progressive uncoupling of oxidative phosphorylation of the kidney mitochondria, a functional abnormality demonstrable also with magnesium deficiency (Vitale et al., 1957b; Skou, 1962).
13.3. Intensification of Magnesium Deficiency and Renal Damage by Excess Phosphates (Animal)
Diets high in phosphate cause not only bone damage and intensify the cardiovascular lesions of magnesium deficiency but also cause renal damage and calcinosis. Shelling and Asher (1932), who were studying the intensification of vitamin D toxicity by diets high in phosphorus and low in calcium, found that even without any vitamin D supplementation, rats on high P/Ca diets developed hypocalcemia, hyperphosphatemia, and "peppering" of the kidneys with calcium deposits, especially in the corticomedullary zone. When given moderately high vitamin D doses (400 times the antirachitic dose), the rats on high P/Ca intakes developed metastatic calcification and died rapidly, in contrast to the tolerance of much higher doses of vitamin D by rats on a 1:1 P/Ca ratio. Maynard et al. (1958) demonstrated that the severe organ changes of magnesium deficiency reflect imbalance among magnesium, calcium, and phosphorus. The diets that produced the highest blood levels of calcium and phosphorus and the lowest blood levels of magnesium caused the greatest renal damage and calcinosis. Forbes (1963) showed that rats fed diets high in phosphorus but low not only in magnesium but in calcium had the greatest degree of renal calcification, even more than the magnesium-deficient rats fed diets high both in calcium and phosphorus. Dunce et al. (1965) also demonstrated that the renal calcinosis of magnesium-deficient rats was aggravated by increasing the dietary phosphate. They also found that increasing the magnesium intake protected against renal calcification. Spaulding and Walser (1970), concerned about the use of high-dosage phosphate therapy in hypercalcemia, administered amounts of phosphate equivalent to those used clinically to rats with hypercalcemia from hypervitaminosis D. They showed that the phosphate clearly increased calcium deposition in kidneys and heart.
Calves fed a magnesium-deficient diet that was not high in calcium but that was relatively high in phosphorus had renal interstitial fibrosis, with some fibrosis of Bowman's capsule; 7 of the 21 calves had marked tubular necrosis, usually with deposits of calcium (L. A. Moore et al., 1938). Comparable lesions were seen in cows with cardiovascular and articular damage associated with a conditioned magnesium deficiency (Arnold and Fincham, 1950).
The marked susceptibility of a strain of mice with hereditary diabetes to cardiac and renal calcification when fed a diet with a high phosphorus/magnesium ratio (1.2/0.04% of diet), and a phosphorus/calcium ratio of 1, for as little as 10 days (Hamuro et al., 1970) is an intensification and acceleration of the changes that develop later spontaneously in this strain. The degree of calcification was little affected by lowering the calcium intake, but was reduced by increasing the magnesium intake to 0.24% of the diet. It was prevented by increasing the magnesium intake to 0.8% of the diet.
Rats with phosphate-mineralocorticoid-cardiac necrosis also have renal calcinosis, and high dosage magnesium is protective (Selye, 1958a,g).
13.4. Mediation by Secondary Hyperparathyroidism; Protection by Parathyroidectomy
The possible source of the mucoprotein that provides the matrix for calcium deposition in magnesium-deficient animals (Bunce and Bloomer, 1972; Dunce and King, 1976/1980) in bone is suggested by the work of Engel (1952). They showed that administration of parathyroid extract resulted in depolymerization of glycoprotein ground substance of bones and cartilage, and deposition of glycoprotein granules in the renal tubules. Bradford et al. (1962) confirmed that rats given parathyroid extract exhibited deposition of intraluminal glycoprotein material, which preceded calcification. Heaton and Anderson (1965) considered the renal cellular damage and calcification of their magnesium-deficient rats (which were fed a diet containing 590 mg of calcium, 440 mg of phosphorus, and 0.3 mg of magnesium/l00 g to be due to secondary hyperparathyroidism, as a result of the magnesium depletion. Parathyroidectomy prevented the renal calcification caused by magnesium deficiency (Heaton and Anderson, 1965), just as it did that caused by phosphate loading (Clark and Rivera-Cordera, 1972b), which also causes nutritional hyperparathyroidism (Clark and Rivera-Cordera, 1972a; Krook et al., 1975; Review: Clark, 1977). Selye (1958c) showed that PTH, in combination with NaH2PO4 caused intense nephrocalcinosis, as well as cardiovascular and bone damage; magnesium chloride was protective against all three experimental lesions.
13.5. Tissue Magnesium Loss and Damage: Not Parathyroid-Mediated
Exploring the mechanisms by which the phosphate-steroid-cardiorenal damage is experimentally produced, and which was first attributed to mediation by hyperparathyroidism, Lehr (1965b) found that sodium phosphate loading of parathyroidectomized rats caused cardiorenal damage even more rapidly than it did in intact rats. In fact, PTH was protective in this model (Lehr et al., 1967). As his group has demonstrated for the cardiovascular system, the sodium phosphate-loading causes tissue magnesium loss and tissue damage, which precedes the rapid induction of renal calcinosis. These animals succumbed with hypocalcemic tetany, cardiorenal necrosis, and calcinosis. Thus, even though comparable lesions could be produced by calcemic agents such as vitamin D or dihydrotachysterol, in the absence of parathyroid glands, the hypercalcemia was not the cause of the lesions. As is suggested by the magnesium-deficiency studies, soft tissue calcification occurs in damaged recipient sites. Lehr et al. (1966, 1967) concluded that depletion of cellular magnesium, however induced, might be involved in initiation of cellular injury, necrosis of both heart and kidneys following demonstrated sharp drops in magnesium levels in both organs of parathyroidectomized sodium phosphate-loaded rats (Lehr et al., 1966). This pharmacologic model is useful in demonstrating the common denominator in dissimilarly caused cardiorenal damage, cellular magnesium depletion. The nature of the lesions, and the sites at which they occur probably depend upon factors such as concomitant hypercalcemia or hyperphosphatemia, levels of local enzymes or mineralization-inhibitors, and physicochemical factors such as pH and the influence of high concentrations of the minerals involved in precipitation of calcium crystals.
13.6. Phosphatases and Extraskeletal Mineralization
Alkaline and pyrophosphatases have been found, not only in bone, where they function to increase mineralization by breaking down the pyro- and other polyphosphates that inhibit mineralization, but also in normal soft tissues, including the cardiovascular system, liver, brain, and kidneys (Gomori, 1941; Kabat, 1941; Kabat and Furth, 1941; Zetterström, 1951; Kirk, 1959; Kunitz and Robbins, 1966; Romanul and Bannister, 1962) and in urine (Fleisch and Bisaz, 1962b). Avioli et al. (1965) noted that elevated urine pyrophosphate levels characterize rapid bone turn over or breakdown, paralleling hydroxyproline outputs. This compound inhibits crystallization of calcium phosphate as apatites. Thus, an increase in its concentration in urine of patients with osteolysis sheds light on McGeown's (1969) report that the evidence of kidney stones is inversely related to that of osteoporosis.
Low levels of activity of pyro- or alkaline phosphatase should diminish the breakdown of these calcification inhibitors. The inhibition of pyrophosphatase by calcium (Kunitz and Robbins, 1966) might explain the paradoxical finding that there was less calcium deposition in kidneys of magnesium-deficient rats, loaded with phosphate and calcium, than there was in those in which the calcium intake was low (Forbes, 1963). It also helps to understand the protection against renal calcinosis of magnesium-deficient rats by calcium administration (Rayssiguier and Larvor, 1973, 1974); and the observations of Hamuro et al. (1970), who fed a strain of diabetic mice diets with different contents of calcium, phosphorus, and magnesium. The mice low in all these elements had more renal and cardiac calcification than did the magnesium/phosphorus-deficient mice on a normal calcium intake. Whether this reflects calcium inhibition of tissue phosphatase is speculative. In a subsequent study, in which the test mice were fed diets low in magnesium and phosphorus, but adequate in calcium, the plasma alkaline phosphatase levels fell from the high levels seen in control diabetic magnesium-supplemented mice (Hamuro, 1971). At the time the low plasma enzyme levels were obtained, there was renal and cardiac calcinosis, a surprising finding, unless the plasma levels are not indicative of the soft tissue levels. It must be noted that these diabetic mice, which have higher plasma alkaline phosphatase levels on the stock diet than do control nondiabetic mice, spontaneously develop calcinosis, although much more slowly than when they are magnesium depleted.
Manifestly, the degree of stimulation of inhibitors of alkaline or pyrophosphatase levels by high magnesium or calcium levels, respectively, cannot be the entire story. The cellular and membrane damage caused by magnesium depletion allows for an intracellular uptake of excess calcium, with deposition of calcium phosphate (usually amorphous but sometimes crystalline) in the damaged cells. Also, patients with hypercalcemia are prone to metastatic calcification despite pyrophosphatase inhibition by calcium. Formation of calcium pyrophosphate dihydrate crystals, such as have been identified in joints might negate the inhibition by pyrophosphate of calcium salt precipitation, when there is hypercalcemia.
13.7 Magnesium Effect on Precipitation of Calcium Crystals in Urine
There are complex interrelations that determine whether or not urine crystals will form in the renal parenchyma or urine. For example, urine containing pyrophosphate has been shown both to inhibit crystallization of calcium oxalate and hydroxyapatites of calcium phosphate (Fleisch and Bisaz, 1962a; R. G. Russel et al., 1964), and to increase the formation of calcium oxalate (Review: Finlayson, 1974). Mucopolysaccharides have been shown to provide the nidus for calcium precipitation in magnesium deficiency and conditions that enhance osteolysis, and to inhibit aggregation and growth of calcium oxalate crystals (W. G. Robertson et al., 1973). Another inconsistency is the hypercalcemia produced by hypervitaminosis D or hyperparathyroidism, which usually is not associated with urolithiasis. Kushner (1956) noted that both conditions cause increased citrate levels, and concluded that citrate-complexing of urinary calcium functions to prevent urolithiasis. There are many other factors that influence susceptibility to stone formation. Only data directly referable to magnesium are considered, briefly, here.
It has long been known that increased concentration of magnesium in the urine increases the solubility of calcium oxalate (Hammarsten, 1929). Rats fed magnesium-deficient diets, which were rich in oxalates and which produced an alkaline urine, had a high incidence of renal calcification and bladder stones; providing a balanced diet without a high Ca/Mg ratio both prevented stone formation and solubilized some that had been formed (Hammarsten, 1938). Mukai and Howard (1963) showed that addition of magnesium to urine of stone-forming patients blocked the ability of such urine to induce mineralization of collagen in vitro. Administration of about 100 mg of magnesium (as the oxide) three times daily, to 11 patients, with recurrent calcium oxalate crystalluria and stone formation, eliminated the crystal formation, although the oxalate was still being excreted, to a lesser degree. The investigators surmised that the magnesium interfered with formation of the crystals. C. Moore and Bunce (1964) found that administration of 420mg of magnesium oxide daily prevented idiopathic hypercalciuria and stone formation and passage in two subjects within two weeks of starting the treatment. One had formed calcium oxalate stones and one had formed calcium phosphate stones. One had the magnesium therapy discontinued after freedom from calculi for five months, and again began passing stones within two weeks. Prien (1965), on the basis of Gershoff's (1959) work indicating the role of pyridoxine deficiency in oxalate formation, included supplementation with 10 mg of pyridoxine hydrochloride with 4 tablets of magnesium hydroxide (providing about 400 mg Mg/day) in his treatment of calcium oxalate stone-formers. Most of his series of 50 patients showed a marked reduction in formation of new stones. Gershoff and Prien (1967) discussed the mechanisms that might be involved in the increased solubility of the calcium salt excreted, the oxalate of which was only moderately reduced, and the calcium of which was actually increased by 25% in patients treated for a year. Of 36 patients who were observed on treatment for 5 years, 30 had no recurrence or decreased incidence of stone formation. They consider the possibility that increased urinary citrate of magnesium-treated patients (that had been low in the stone formers) might be contributory to the increased solubility of calcium. Melnick et al. (1971/1973; 1971) have reported similarly favorable results among 95 recurrent calcium oxalate stone formers treated with 100 mg of magnesium as the oxide twice daily for two years, and among 47 treated for 4 years. J. Thomas et al. (1978) have demonstrated in vitro and in vivo that magnesium inhibits formation of calcium oxalate crystals. They have obtained the best clinical results with use of magnesium trisilicate, providing 300 mg of magnesium daily. It may be this simple physicochemical effect that is responsible for the difference in incidence of urolithiasis in hard- and soft-water areas (pp. 21-24).
13.8. Clinical Renal Diseases Possibly Related to Magnesium Deficiency
The experimental evidence that magnesium deficiency during pregnancy produces greater fetal than maternal magnesium deficiency raises the possibility that renal tubular abnormalities, such as are produced in weanling magnesium-deficient animals, might occur in utero. No studies of the renal structure of fetuses of experimental magnesium-deficient mothers have been found, and thus this possibility remains speculative. Microscopic examination of kidneys of stillborn babies of mothers subject to magnesium deficiency should provide valuable data.
After birth, there are several conditions that lead to magnesium deficiency, both in the neonatal period and later in infancy. Infants who do not survive neonatal asphyxia or sodium bicarbonate or sodium lactate treatment of their neonatal or postoperative acidosis, both anoxia and acidosis causing egress of magnesium from the cells, should have their renal parenchyma carefully studied, especially for evidence of tubular cellular and subcellular damage. The kidneys of erythroblastotic infants failing to survive exchange transfusion with citrated blood (which chelates magnesium) should be similarly examined. Definitive data can be obtained from experimental models of these perinatal abnormalities, which can provide electron microscopic evidence of very early renal changes, as well as light microscopic evidence of sequellae. Such studies should include evaluation, not only of kidneys, but of cardiovascular tissues (especially intramural coronary arteries, myocardium, and endocardium) and bone.
Because neonatal hypocalcemia is usually noted first, and usually aggressively treated with calcemic agents to control the neuromuscular irritability and convulsions, what might be an underlying magnesium deficiency is usually detected only by the time magnesium depletion has developed to the point of severe hypomagnesemia. Familial magnesium malabsorption might be a contributory factor in infants and children with the most severe manifestations. Neonatal magnesium deficiency and hypoparathyroidism secondary to gestational magnesium deficiency in some instances, and to high phosphate + vitamin D intakes in others, is likely to contribute to less severe but possibly damaging tissue magnesium loss. The renal damage of such treatment might lead to long-term intensification of magnesium deficiency by causing damage to the portion of the renal tubules where active magnesium reabsorption occurs.
13.8.1. Renal Tubular Defects in Magnesium Reabsorption
As noted earlier, most active renal tubular reabsorption of magnesium occurs in the ascending limb of the loop of Henle (ALLH), which provides insight into clinical magnesium wastage, most early experimental magnesium deficient renal damage occurring in the tubular cells of the corticomedullary area, with microliths of the loop of Henle, convoluted and distal tubules, and with damage to ALLH. That these experimental findings are relevant to the clinical situation is suggested by fragmentary findings.
Whether magnesium deficiency contributes to clinical aminoaciduria, as it does the experimental model should be investigated. Its occurrence in renal tubular acidosis, vitamin-D-resistant rickets, and hyperreactivity to vitamin D, in all of which conditions magnesium deficiency might play a role, is suggestive.
13.8.1.1. Contributions to Clinical Renal Magnesium Wastage by Calcemic Factors and Phosphate Therapy
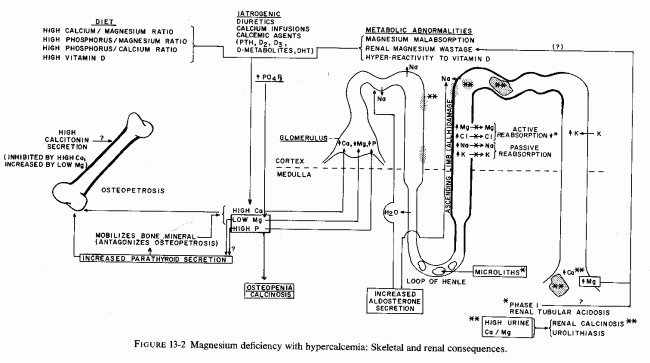
Calcium deposits in the lumens of the proximal renal tubules and of the ALLH have been described in an infant whose symptomatic hypocalcemia had been unsuccessfully treated with calcium infusions and high-dosage vitamin D, before severe magnesium deficiency was detected in a specimen taken the last day of life (Vainsel et al., 1970). Another infant, whose persistent infantile hypocalcemia had been treated by intensive calcium therapy and then, when complicated by intractable diarrhea, with addition of high-dosage vitamin D (10,000 IU/day) was then found to have hypomagnesemia and tubular acidosis. At autopsy there were calcium deposits in the distal tubules and collecting ducts. No parathyroid tissue was found at autopsy (Taitz et al., 1966). Infants with hypervitaminosis D and infantile hypercalcemia, which seems to be caused by hyperreactivity to vitamin D (Review: Seelig, 1969b) have also been found to have intraluminal calcium deposits predominantly in the outer half of the renal medulla (Dawson et al., 1954; Rhaney and Mitchell, 1956). Nephrocalcinosis infantum, that Lightwood (1935) first associated with renal tubular acidosis, hypophosphatasia, hyperoxalemia, and sarcoidosis (which is associated with hyperreactivity to vitamin D) is also associated with renal tubular lesions and calcium deposits involving Henle's loop and tubules immediately proximal and distal to it (J. A. James, 1956; Kushner, 1956; Shanks and MacDonald, 1959; T. Ferris et al., 1961; Paunier et al., 1968a). Hyperreactivity to vitamin D, of infants who were receiving excessively fortified milk and infant foods was implicated in Great Britain in renal tubular acidosis (Fig. 13-2) (Lightwood and Butler, 1963) and in nephrocalcinosis (Shanks and MacDonald, 1959). Development of such abnormalities in children who were being given high-dosage vitamin D for vitamin D-refractory rickets (with or without aminoaciduria) or for idiopathic hypoparathyroidism and hypocalcemia (T. Ferris et al., 1961; Paunier et al., 1968a; Moncrieff and Chance, 1969) suggests that magnesium deficiency might have played a contributory role, first to the vitamin-D-refractory rickets or hypocalcemia and then to the increased susceptibility to nephrotoxicity of vitamin D. It seems plausible that the presenting tetany and convulsions of the babies with hypocalcemia, renal tubular acidosis, and nephrocalcinosis (J. James, 1956; Ferris et al., 1961) might have been contributed to by magnesium deficiency. Familial renal tubular wasting of magnesium has been reported in siblings with renal tubular acidosis, renal calcinosis, and hypocalcemia that was resistant to very high doses of vitamin D (Michelis et al., 1972). The older child (a 10-year-old girl) had active rickets; the bone age of the younger brother (6 years old) was three standard deviations below normal. Neither child's hypomagnesemia (1.1, 1.2 mEq/liter) responded to high- dosage oral magnesium supplementation, a failure found due to renal wastage rather than malabsorption of magnesium. It is conceivable that the initiating abnormality in these children might have been hyperreactivity to vitamin D, which might have led to hypercalcemia and ALLH damage, that was responsible for their persistent renal magnesium wastage. It is provocative, however, that both children had been born prematurely. The older child was jaundiced at birth and developed a convulsive disorder at the age of 6. The younger child underwent surgery at 6 weeks of age. The premature births and complicated neonatal courses might have contributed to early magnesium deficiency that might have contributed to hyperreactivity to (toxic effects of) vitamin D. The older child had had polyuria from the age of two; the younger child from the age of 1.
Another child with vitamin-D-resistant rickets was found to have hypomagnesemia (0.7 mEq/liter) and two times the normal renal clearance of magnesium when he was ten years old, following two years of very high (15 mg/day) vitamin D dosage (Sann et al., 1975). He had manifestations similar to those seen in infantile hypercalcemia during the first year of life (anorexia and insomnia), and he exhibited poor growth. He had clear evidence of kidney disease from two years of age, at which time he developed proteinuria; by six he had polyuria and polydipsia and marginally high serum calcium. By eight he had skeletal demineralization, for which the high-dosage vitamin D was prescribed. Renal biopsy showed juxta-glomerular hyperplasia, and he had hyperrenism and normotensive aldosteronism. At this time, sodium restriction caused hyponatremia. Angiotensin-infusion did not raise his blood pressure, and he was judged to have a form of Bartter's syndrome (Bartter, 1962). A second renal biopsy confirmed the juxta-glomerular hyperplasia and revealed proliferative endarteritis of the efferent arterioles. Two years later, the osteoporosis had progressed, and his phosphaturia, magnesiuria, and hypophosphatemia and hypomagnesemia were identified and found to be refractory to the potassium-sparing diuretic (triamterene) that was given in the hope that it might spare magnesium. It is provocative that hypomagnesemia has also been identified in Bartter's syndrome (Brackett et al., 1968; Sutherland et al., 1970; Mace et al., 1973), a condition that Kurtzman and Gutierrez (1975) suggest may be a syndrome caused by ALLH dysfunction.
Reviews of the literature on renal damage caused by hypercalcemia-inducing agents such as vitamin D excess (Epstein, 1960) and hyperparathyroidism (Pyrah et al., 1966) show that the earliest lesions are in the loop of Henle, the ALLH, and the distal convoluted tubules and collecting tubules and that the duration of the hypercalcemia can be as short as one to three days to produce significant damage. The damage becomes irreversible with sustained hypercalcemia. That such lesions can cause renal magnesium loss is suggested by the work of Massry et al. (1967), who reported an acquired defect in renal tubular reabsorption of magnesium in a 44-year- old woman with surgical hypoparathyroidism. They considered the renal defect as one likely to have been caused by hypercalcemia, subsequent to long-term treatment with vitamin D and thiazide diuretics (each of which favors retention of calcium over magnesium). Hypomagnesemia was detected (0.9-1.2 mEq/liter), which was not corrected by withdrawal of the diuretic and the vitamin D. Her high renal clearance of magnesium persisted, and did not change on administration of PTH. Hypercalcemia of immobilization of an adolescent boy, who had suffered multiple fractures, also resulted in sustained increased renal clearance of magnesium (Hyman et al., 1972).
It is possible that early renal tubular damage, caused by calcemic agents (supra vide) or by phosphate therapy that causes soft tissue calcinosis, including renal tubular damage (Bulger et al., 1930; Albright et al., 1932; Carey et al., 1968; Marti and Cox, 1970; Dudley and Blackburn, 1970) can be reversed with adequate magnesium repletion, once the hypercalcemia has been controlled. A clue that this may be so derives from the observations of Dooling and Stern (1967), who studied the magnesium status of a six-day-old infant whose hypocalcemic convulsions had been intensively treated with oral and parenteral calcium. (It should be kept in mind that such infants are also generally hyperphosphatemic. The baby excreted large amounts of magnesium (16.3 mg/kg/24 hr) while he was hypomagnesemic (0.6-0.7 mEq/liter) and being treated by intramuscular administration of 0.25 ml of 50% magnesium sulfate every 6 hours (25 mEq M2+/24 hr). His urinary magnesium output did not fall until the magnesium dosage was doubled. By the fourth day of high- dosage magnesium, with resultant stable elevation of his serum magnesium level at 1.6 mEq/liter, his urinary magnesium output had fallen to 3.06 mg/kg/24 hr, a fivefold drop in daily urinary magnesium output. It is tempting to speculate that microliths might have formed in the loops of Henle, with resultant early damage to the broad ascending limb when he was being loaded with calcium and was magnesium depleted. Early during the magnesium supplementation, there might have been impaired tubular reabsorption of magnesium. When the amount of magnesium given was increased, solubilization of presumed calcium microliths and tubular cells might have taken place, with resultant increased active tubular reabsorption of magnesium.
13.8.1.2. Contribution to Clinical Renal Magnesium Wastage by Malabsorption
Specific genetic magnesium malabsorption or general intestinal malabsorption might cause severe enough magnesium depletion to cause renal tubular damage directly, or as a consequence of calcemic therapy of secondary hypocalcemia. Direct evidence that this might be so has recently been provided by Rapado and his colleagues (Rapado et al., 1975; Rapado and Castrillo, 1976/1980a). They reported a 12-year-old child with a diagnosis of nephrocalcinosis from early life, who developed overt rickets when her hypercalciuria was treated, first with sodium cellulose phosphate and then with hydrochlorthiazide. She was then found to have hypomagnesemia (0.5 mEq/liter) and required parenteral magnesium therapy because she was both a magnesium malabsorber and renal waster. Of two additional patients with nephrocalcinosis, one young man with latent tetany of hypomagnesemia (0.75 mEq/liter) and hypocalciuria (6.8 mg/100 ml), reported by this group, was a renal magnesium waster. Their third patient with hypomagnesemic (0.65 mEq/liter) nephrocalcinosis was a young man whose urinary magnesium levels did not decrease on a low-magnesium intake, and whose fecal excretion of magnesium was higher than his intake, suggesting magnesium malabsorption. This patient's serum calcium level was only marginally low (8.3 mg/100 ml), and he had tachycardia and hypertension. All three patients had hypercalciuria, and their urinary calcium output increased with magnesium therapy.
The patient reported by Freeman and Pearson (1966) as a renal tubular waster of magnesium, had had a history suggestive of steatorrhea and growth failure during her first year of life, which suggested early magnesium depletion. She came from a family with a high incidence of hypomagnesemia: One of her sons had serum magnesium levels of 1.12, 1.20 mEq/liter on two occasions; another son, a sister, a maternal cousin, a daughter, and the patient's mother had marginally low serum Mg levels (1.50, 1.47, 1.68, 1.51, and 1.70 mEq/liter, respectively, strongly suggestive of a genetic trait. Whether the inherited trait was primary renal dysfunction or magnesium malabsorption is not clear. Another patient, one who had had gastrointestinal disease (ascariasis, followed by gastroenteritis) before she was two years old, exhibited subsequent growth failure, intermittent glycosuria and aminoaciduria, and X-rays suggestive of osteoporosis (B. Booth and Johanson, 1974). The boy, whose hypomagnesemia was not detected until he was six years of age (Miller, 1944), might have had infantile hypomagnesemia, as suggested by his long history of neuromuscular irritability. His osteochondritis of three years duration, resembling that of a five-year-old boy with renal magnesium wasting (Klingberg, 1970), suggested to Booth and Johanson (1974) that the child reported by J. F. Miller (1944) might also have had a renal tubular defect. It should be noted, however, that the defect might as readily have been in intestinal absorption of magnesium
Perhaps the first recorded instances of excessive urinary output of magnesium despite marked hypomagnesemia were two patients with peptic ulcers, one with prolonged nasogastric suction (who undoubtedly was not absorbing normal amounts of magnesium) and another who had had several complicated surgical procedures (Martin et al., 1952). That surgical patients do not conserve magnesium efficiently during the early postoperative days has been shown repeatedly. Data are generally not available on the nature of the antacids taken, prior to the suction or surgery, but if they were calcium rather than magnesium preparations, the patients might have had a relatively high Ca/Mg dietary ratio prior to the acute situation that intensified their magnesium loss
Note should be taken of the incomplete distal renal tubular acidosis seen in two women who developed hypomagnesemic hypocalcemia as a result of intestinal malabsorption: one following intestinal resection for regional enteritis, the other with nontropical sprue (Passer, 1976). Since these patients had hyperparathyroidism (by immunoassay) and treatment with magnesium alone corrected the abnormal renal function, the author speculated that the resistance to the calcemic effects of the endogenous PTH might be related to abnormalities in vitamin D metabolism, which was corrected by the magnesium.
13.8.1.3. Miscellaneous Factors in Renal Magnesium Wastage
Randall et al. (1959) was the first to propose a functional renal tubular defect as an explanation for the failure of a 38-year-old man with mild diabetes mellitus, pyelonephritis, focal seizures, and electrocardiographic abnormalities to conserve magnesium. Before dying with extensive arteriosclerosis and myocardial infarct, he had exhibited hypokalemic alkalosis, hypocalcemia, and hypophosphatemia. They noted that additional patients with renal tubular disease wasted magnesium. Two adult sisters and an unrelated 22-year-old woman were first identified as having impaired renal conservation of magnesium and potassium, in association with metabolic alkalosis; one also had hypochloremia (Gitelman et al., 1966a). The sisters had dermatologic manifestations resembling those seen in magnesium-deficient animals; the third patient had recurrent carpopedal spasm. All exhibited slightly elevated aldosterone secretion without hypertension and had minor ECG abnormalities; two had muscle weakness.
Chronic hypomagnesemia and recurrent episodes of neuromuscular irritability and severe abdominal pain have recently been attributed to a renal tubular defect in magnesium reabsorption in a boy, whose mother also has had subnormal serum Mg levels (Paunier and Sizonenko, 1976/1980). As in the previously reported patients with this renal defect, even supplementation with large doses of magnesium failed to normalize the serum magnesium levels.
The oldest patient, at the time of first detection of renal wastage of magnesium, is a postmenopausal woman who developed normocalcemic latent tetany of marginal deficiency several years after total hysterectomy (Seelig et al., 1975). She has a less severe form of renal Mg wasting and less marked hypomagnesemia than the other cited patients. Her condition is associated with normotensive, intermittent aldosteronism and increased plasma renin activity (PRA), only manifest in response to dietary Mg restriction or to hormonal challenge (i.e., deoxycorticosterone acetate) that increased her magnesium deficit (Seelig et al., 1976/80). Since she has also had hypochloremia, it has been proposed that she has malfunction of the ALLH, where not only magnesium but chloride is actively reabsorbed (Rocha and Kokko, 1973; Burg and Green, 1973; Kurtzman and Gutierrez, 1975). It seems plausible that her renal dysfunction and concomitant abnormalities-sodium retention, peripheral edema, hypokalemia responsive to magnesium, hypercapneic alkalosis, and hormonal aberrations-may be the result of magnesium insufficiency, since all of these findings have been reported in experimental magnesium deficiency (Review: Seelig et al., 1976/1980; Whang and Welt, 1963; Ginn et al., 1967; Cantin, 1970; Elin et al., l97la; El Shahawy, 1971; Cantin and Huet, 1973).
13.8.2. Renal Damage during Pregnancy: Related to Magnesium Deficiency?
Magnesium deficiency has been implicated in preeclampsia and eclampsia. Possibly it contributes to the renal damage of eclampsia. The involvement of the small (coronary) arteries in magnesium deficiency makes one suspicious that the renal arteriolar disease of young toxemic primiparas (Smythe et al., 1964), who are particularly prone to magnesium deficiency of pregnancy, might also be a consequence of magnesium inadequacy. DeAlvarez and Gabrio (1953) implicated arteriolar spasm in the decreased glomerular filtration rate of patients with toxemias of pregnancy.
The attempts to counter the leg cramps of pregnancy (which might be contributed to by magnesium deficiency) by calcemic therapy, might intensify the magnesium deficiency directly and as a result of damage to renal tubular cells (supra vide). The resultant high calcium/magnesium ratio particularly in arterial tissue [such as has been implicated in increased arterial tension (Review: Haddy and Seelig, 1976/ 1980)] might similarly be a factor in the hypertension of abnormal pregnancy. Calcemic supplements to magnesium-deficient pregnant women might contribute to urinary calculi of pregnancy, which has reported in 0.05-0.35% of pregnancies. (McVann, 1964; R. Harris and Dunnihoo, 1967). Since estrogen lowers the urinary content of calcium and raises its citrate level (Shorr, 1945), both effects that militate against calcium stone formation, the degree of magnesium deficiency might well be fairly profound for calcareous stones to form during pregnancy. On the other hand, the resultant hyperparathyroidism of pregnancy might directly increase the propensity toward renal calcinosis formation, as well as hypertension, both being consequences of hyperparathyroidism (Review: Pyrah et al., 1966).
13.8.3. Diabetic Renal Disease: Contributed to by Magnesium Deficiency?
Only brief reference will be made here to the speculation that magnesium deficiency might be contributory to proliferative arteriolar sclerosis, found in the kidneys as well as in other tissues, including the myocardium. As in experimental magnesium deficiency, in which there is arteriolar disease with subendothelial, muscle wall, and endothelial proliferation, with increased wall thickness/lumen ratio, renal (and other) arterioles have subintimal and medial abnormalities with encroachment on the lumen (Review: Ditzel, 1954). The lesions are not identical pathologically and the glomerular capillary changes of the Kimmelstiel-Wilson lesion have not been described in magnesium deficiency, but loss of magnesium by diabetic patients raises the possibility that the arterial changes might have a component contributed to by magnesium deficiency.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
{kind=link}
{kind=link}