MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part II: Chapter 6
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
6
Is Clinical Arteriosclerosis a Manifestation of Absolute or Conditioned
Magnesium Deficiency?
6.1. The Arterial Wall and Arteriosclerosis
The major emphasis of the preceding sections on magnesium-lipid interrelationships and on estrogen-lipid/coagulation-lipid interrelationships is predominantly on blood constituents, as they influence the development of atherosclerosis.
The likelihood that metabolic and structural alterations in arterial walls may predispose to their increased accumulation of lipids has also been investigated. The need to consider, not only alterations in the constituents of blood but also in the status of the containing vessels, was commented upon by Duff and McMillan (1951) in their review of changing concepts of the pathogenesis of arteriosclerosis. They observed that the view that chemical and physicochemical aberrations of the serum lipids and lipoproteins are fundamental to the pathogenesis of arteriosclerosis had become so popular that "… the casual reader of recent literature might wonder whether some authors conceive of an atherosclerosis so independent of the substrate of the vessel wall, that it may occur in the absence of the blood vessels themselves."
6.1.1. Mucopolysaccharides and Elastica in Arteriosclerotic Arteries
Specific alterations in the mucopolysaccharides have been observed in the ground substance of arteriosclerotic arteries obtained from human material. Increased metachromasia, due to elevation in acid mucopolysaccharides, occurs in arteries from human material prior to lipid infiltration in aging and in arteriosclerosis (Faber, 1949; Moon and Rinehart, 1952; Moon, 1959; Gresham et al., 1962). It has been suggested that it develops in areas characterized by prior degeneration of the elastica and predisposes to infiltration by lipids (Moon and Rinehart, 1952; Taylor, 1953; Moon, 1957, 1959). On the other hand, it has been postulated that lipids in arterial lesions derive from the degenerated elastic fibers and that the elevation in mucopolysaccharide reflects a healing process (Zugibe and Brown, 1960; Zugibe, 1963).
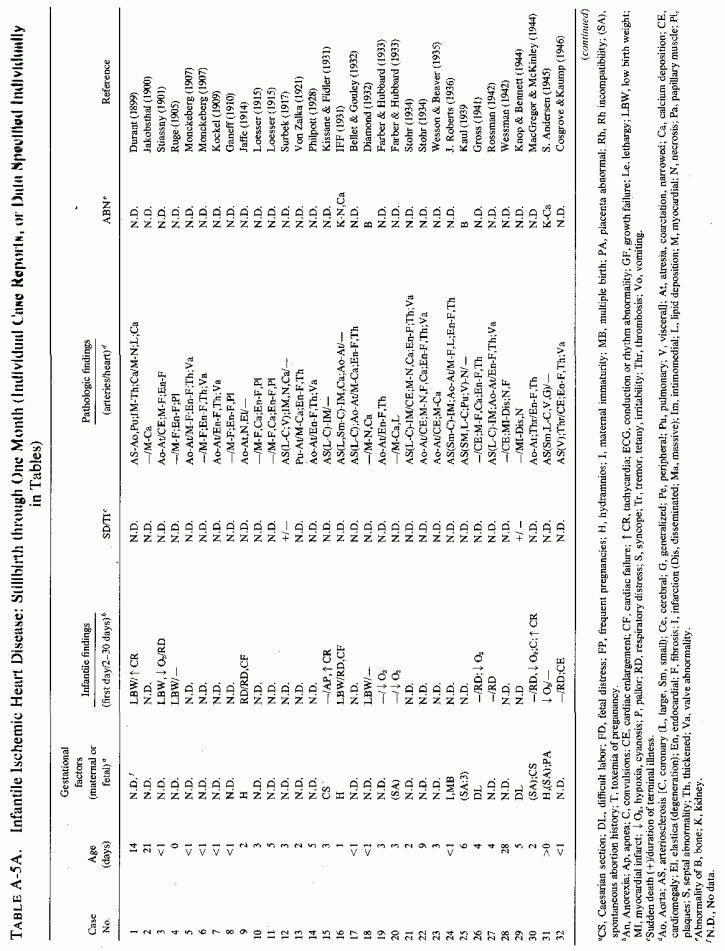
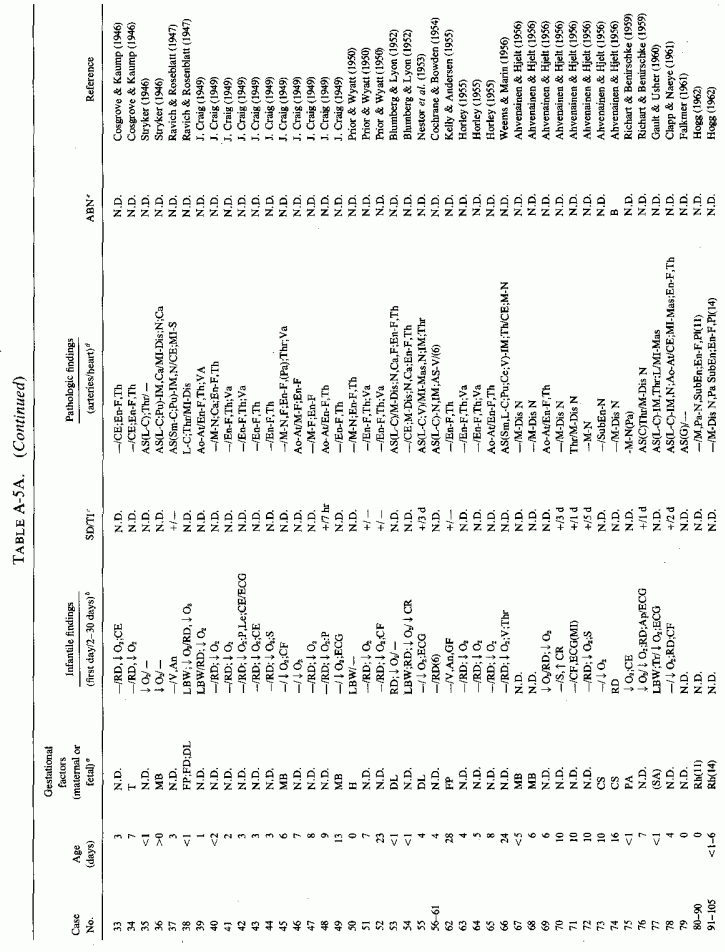
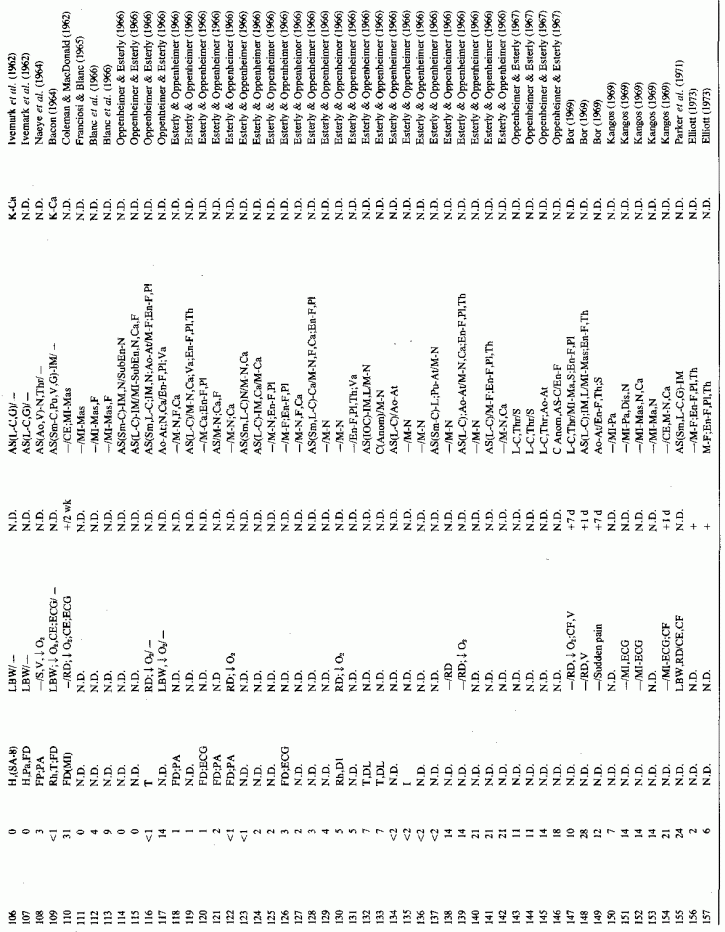
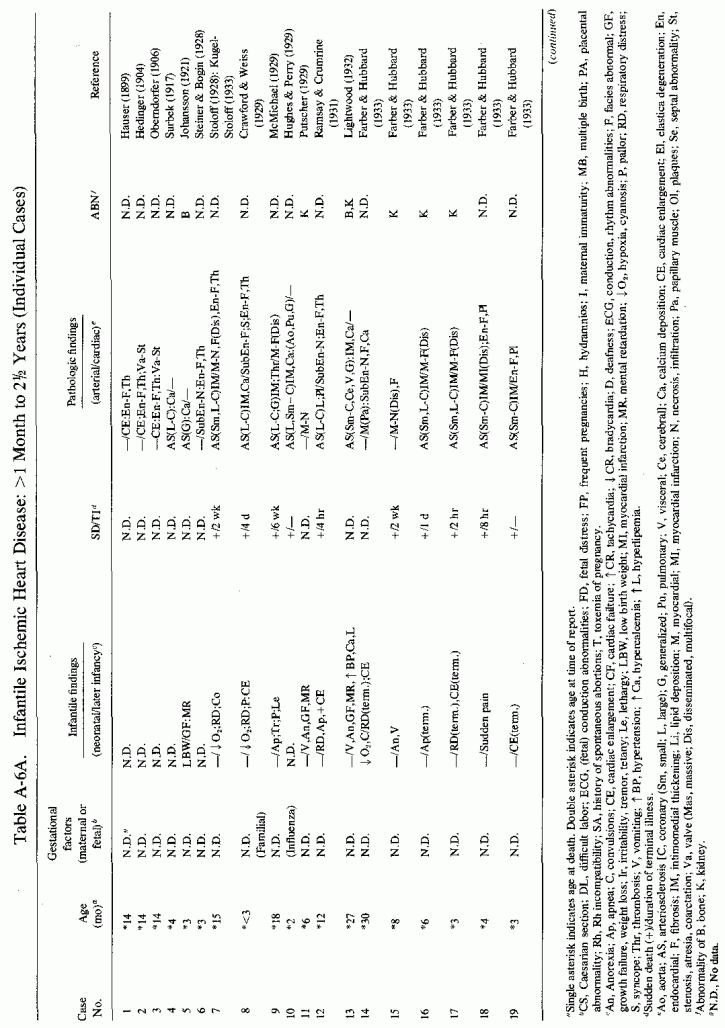
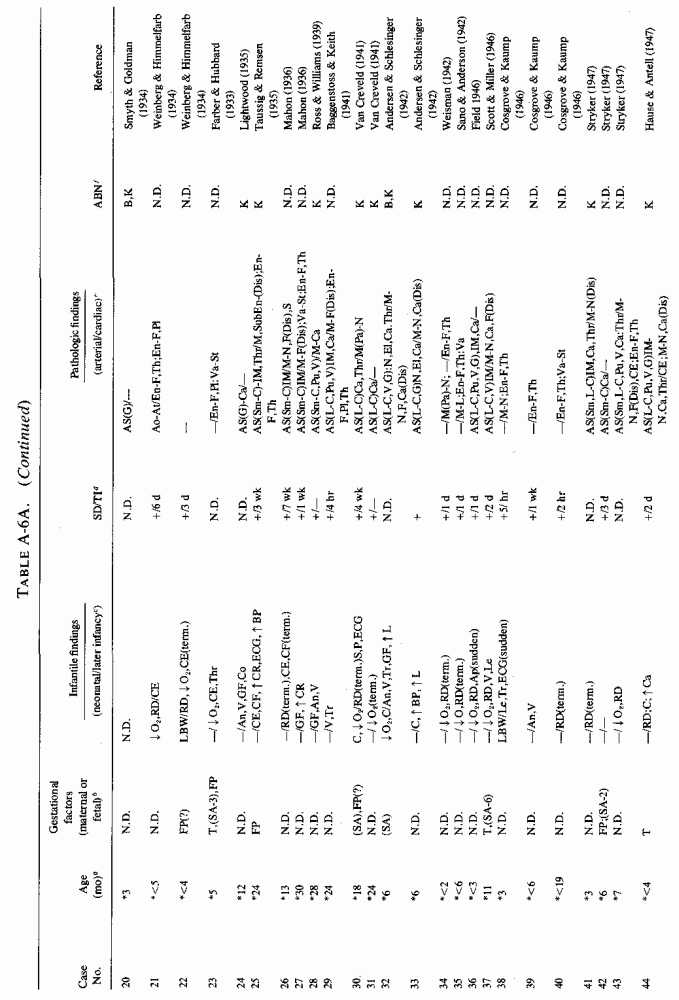
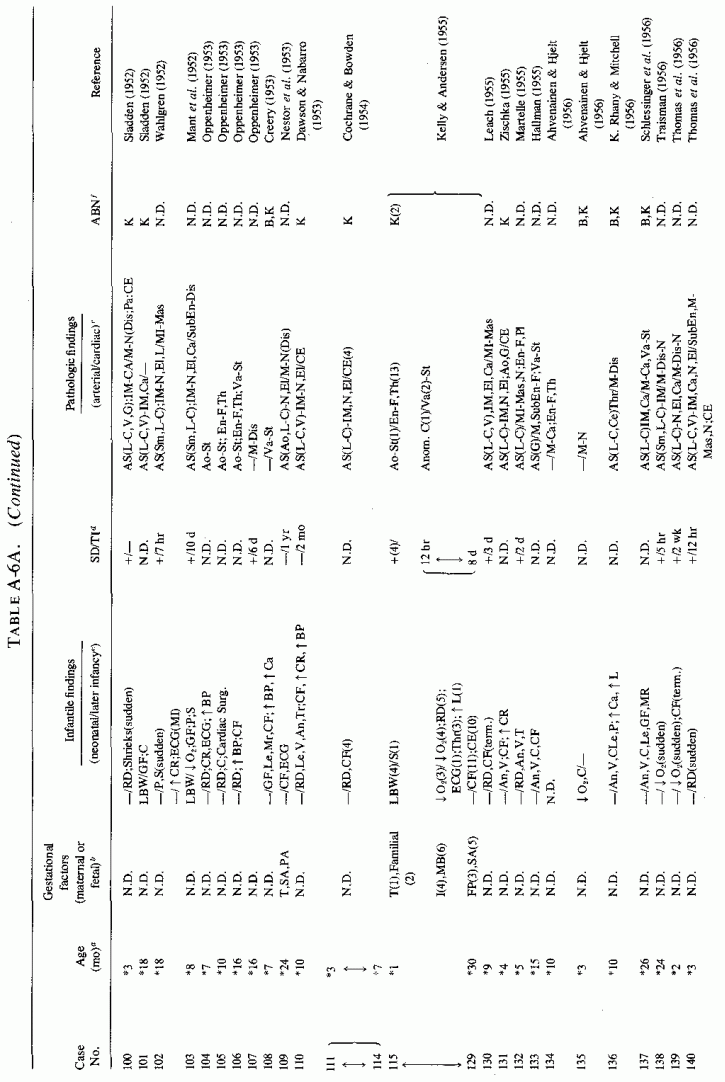
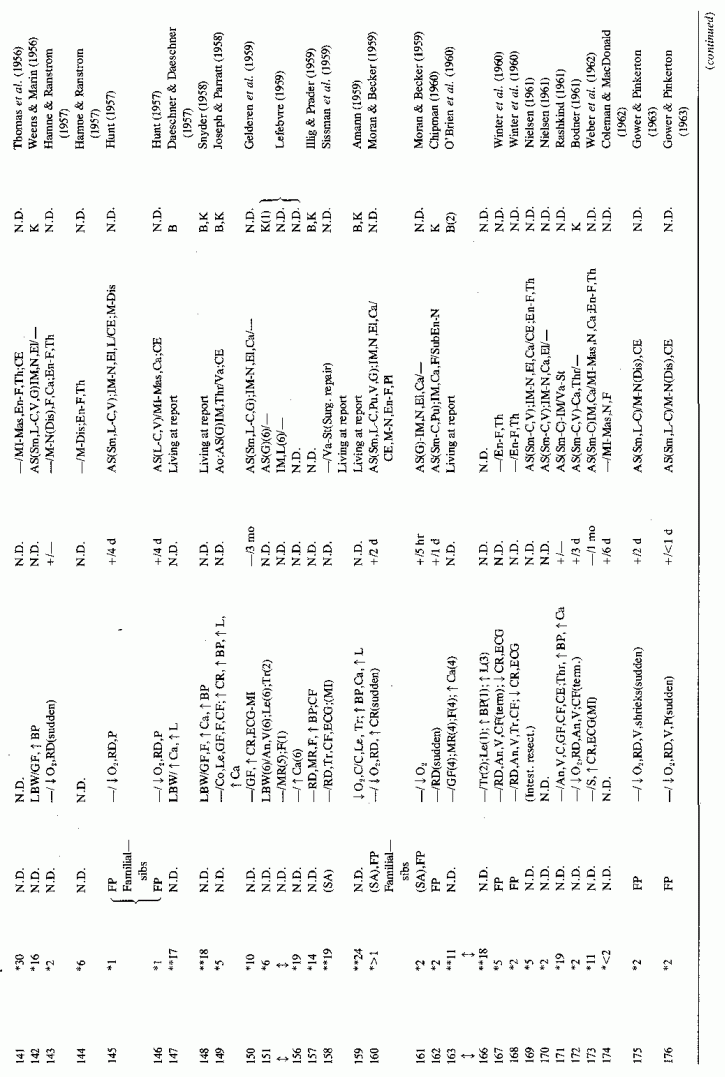
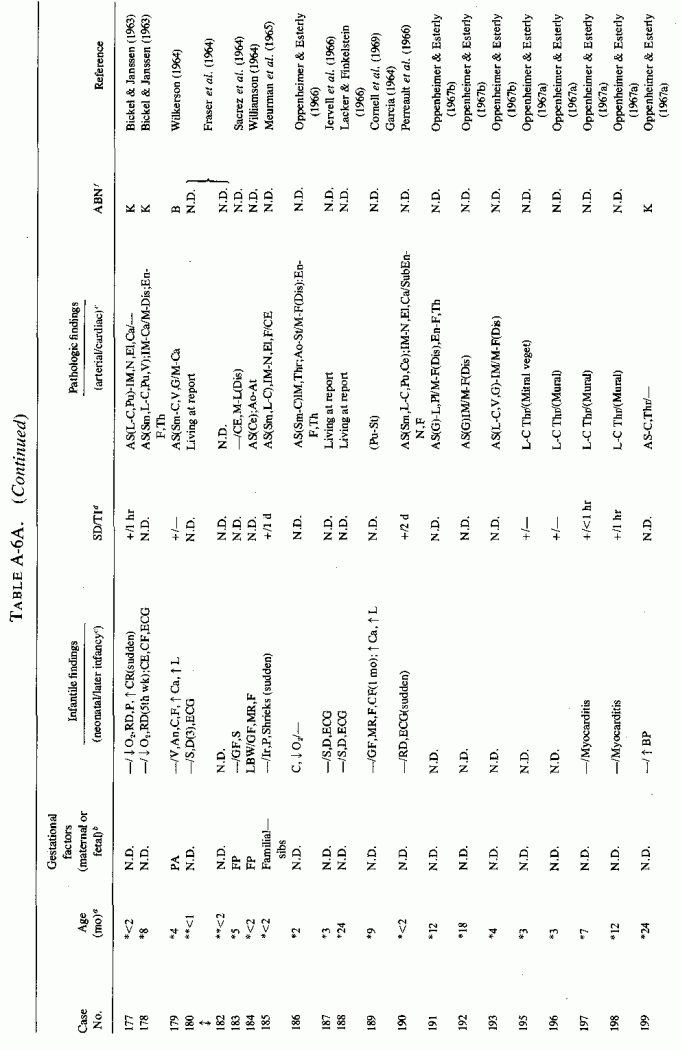
6.1.2. Pathology of Infantile Arteriosclerosis (See Appendix Table A-5A, Table A-5A continued, Table A-5A continued (2), Table A-5B, Table A-6A, Table A-6A continued, Table A-6A continued (2), Table A-6A continued (3), Table A-6A continued (4), Table A-6A continued (5), Table A-6A continued (6), Table A-6B and Table A-6b continued.)
Intimal, subintimal, and medial arterial lesions, usually of the small- and medium-sized arteries, such as have been described in infants who died suddenly or after protracted congenital cardiac disease, are characterized by elastica degenerative processes, mucopolysaccharide or calcium deposition, and proliferative or fibrotic intimal and medial changes. Lipid droplets are often also seen, but the fat deposition does not become atheromatous until later in infancy and childhood. The very early infantile arterial lesions resemble those of magnesium deficiency in animals with otherwise balanced diets, i.e., "pure" magnesium deficiency. Suddenly fatal arterial lesions of infants have usually been coronary (associated with perivascular myocardial microfocal necrosis, or more rarely with gross infarctions). However, most of the infants with coronary lesions also had arteriosclerosis of other viscera and occasionally had generalized arteriosclerosis. Whether earlier arterial lesions exist in infants who develop "adult-onset" atherosclerosis or in infants born to parents with early cardiovascular disease is difficult to ascertain.
Even among infants identified as having had cardiovascular disease pre- or postmortem, the degree and location of arterial damage are often not specified. Among the 157 separately cited cases of infants born dead or dying within the first month of life with cardiac lesions, 30 had coronary arteriosclerosis described and 14 had visceral or generalized arteriosclerosis described. Although not mentioned, coronary arterial lesions are probable in at least 80 more who had myocardial lesions ranging from necrosis with and without calcification to fibrosis. Only 5 of the 80 with endocardial fibroelastosis had coronary arterial lesions mentioned. Among the 253 infants tabulated as having died of cardiovascular disease from 1 month to 2 1/2 years of age, 85 were described as having coronary arteriosclerosis, with or without involvement of other arteries. Myocardial lesions suggestive of ischemic heart disease were described in an additional 74 infants, whose coronary arterial status was not described. Almost half of the 110 infants with endocardial fibroelastosis did not have coronary arterial or myocardial lesions described. Among those whose arterial lesions were described, a third of those up to 1 month of life had intimomedial proliferation and almost as many had thrombosis noted. About half of the infants of 1 month to 2 1/2 years of age, whose arteries were described, had intimomedial proliferation, but only a tenth had thromboses. It is not possible to ascertain the incidence of intimomedial proliferation from surveys of autopsy material, for some include intimal sites of proliferation, "cushions" as precursors of atheromata (Dock, 1946; Fangman and Hellwig, 1947), and others specifically exclude them as normal variants (Schornagel, 1956; Oppenheimer and Esterly, 1967).
With one exception, the 19 children whose arteries showed degenerative or calcific changes were no more than 4 days old at death. This might be supportive of Gruenwald's (1949) conclusion that perinatal hypoxia can cause arterial necrosis, based on his finding such lesions in as many as 9.5% of infants autopsied after stillbirth to 3 days of life. There were fewer instances of intimomedial degenerative changes in the older infants, but more instances of calcification. Three cases of lipid deposition in the arteries were noted in the individual case reports of infants up to one month of age; 6 were noted in the group up to 21/2 years.
Few patients with supra- or subvalvular aortic stenosis or with cardiofacial peculiarities are cited; most survived beyond the 2 1/2-year limit selected. That these children probably developed their abnormality either in utero or in the first 2 years of life seems likely.
6.1.3. Incidence of Infantile Coronary Arteriosclerosis
This is a disease, the incidence of which is impossible to estimate. As a result of the effort to classify infants with histopathologically identical lesions as suffering from different diseases, depending on coexisting anomalies or demonstration of conditions that predispose to metastatic calcification, there is not uniformity of reporting. Further complicating the determination of the incidence of infantile coronary arteriosclerosis is the lack of agreement as to what the infantile arteriosclerotic lesion is. In "idiopathic" infantile arteriosclerosis, intimal thickening and elastica degeneration are recognized as the typical findings, but focal intimal proliferative, termed "cushions" (usually with fibromuscular disorganization of the media), which are found more than twice as often as are atheromas, are not uniformly considered pathological. When only atheromatous lesions are considered evidence of arterial disease, neonatal focal myocardial necrosis has been reported in the absence of lesions of the main coronary arteries. Rarely are the intramyocardial arteries examined. Thus, coronary occlusion or significant coronary disease is less frequently reported than is that of the myocardium or endocardium. Nonetheless, an attempt to select, from the pathology surveys, cases designated by the age groups selected here, and that exclude major anomalies (other than atresia of the great vessels), suggests about 500 in which hypoxia of the heart might have been involved in infants up to one month of age, and over 2000 in those from 1 month to 2 1/2 years of age.
In the case of endocardial fibroelastosis (EFE), myocardial ischemia has been repeatedly implicated. J. M. Craig (1949), who presented 43 cases, noted that microscopic myocardial necrosis and fibrosis was common. He suggested intramural coronary disease in utero as a contributory factor. F.R. Johnson (1952) suggested that intrauterine anoxia might be contributory to the EFE seen in malformed hearts; Moller et al. (1966) noted that infarcts of the papillary muscles are not infrequently found in infants with EFE. Since the subendocardial myocardium obtains oxygen from the blood in the heart chambers, conditions that interfere with blood outflow, that lead to stagnation, can lead to hypoxic subendocardial and endocardial damage and thickening. In fact, outflow obstruction is the most common anatomic disorder associated with EFE (Moller et al. 1964; Bryan and Oppenheimer, 1969).
A survey of necropsy material in a major medical center showed that myocardial infarction is not rare in infants, even occurring in utero (Franciosi and Blanc, 1968). In infants with congenital heart disease, the infarcts were limited to papillary muscles (which are supplied by the small end-arterial branches of the coronaries), and to microscopic lesions of the subendocardial ventricular myocardium that were adjacent to perivascular and interstitial fibroses. Although none was associated with occlusive arterial disease, grade 1 to 4 coronary lesions were found frequently. Grade I was characterized by frayed intimal elastica lamina; grade 2 additionally had slight focal intimal fibrosis; grade 3 had intimal cushions in addition; grade 4 had diffuse elastica fraying and diffuse intimal thickening equaling the thickness of the media. The frequency of the infantile myocardial infarcts was 80% among those with anomalous venous return, 89% in those with pulmonary valvular stenosis, and 100% in those with aortic valvular stenosis.
The coronaries are often not examined, even among infants who die during the perinatal period and are autopsied. This is particularly so in the case of the small- and medium-sized arteries, which are most often involved in infantile coronary arteriosclerosis, and which are most likely to be involved in focal and microscopic myocardial necrosis and fibrosis and in fibroelastosis. Blanc et al. (1966) pointed out that systematic examination of the small- to medium-sized coronaries of infants has disclosed that as many as 12% had arteriosclerosis.
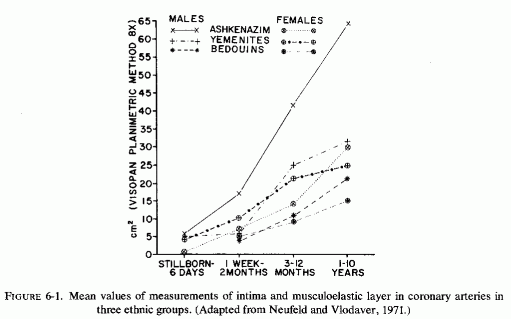
Despite the fact that many of the infants with necropsy evidence of coronary disease had died suddenly, none were recorded as having been reported by medical examiners or coroners (Moran and Becker, 1959). Thus, it seems likely that many of the instances of this disease are not recognized. Supporting the contention that many cases might be missed are the studies of autopsy material that include examination of the large coronaries of infants. In a study of the proximal segments of the main coronaries of 105 individuals who died before birth to the early twenties, only the fetuses (24 of 3½-9 months gestation) were free of coronary lesions (Moon, 1957). In that series, two premature infants had ruptured internal elastic membranes but had no other coronary lesions. Most of the 52 infants under two years of age had coronary lesions, the earliest noted being rupture and degeneration of the internal elastic membrane. Some also had fibroblastic proliferation with deposition of mucopolysaccharides and proliferation of endothelial cells overlying these areas. Infants several months old had progression of the intimal lesions as compared with newborn infants, the intimal thickening being very pronounced at three or four months of age. The intima was commonly thicker than the media. Serial sections of the left anterior descending coronaries of 88 infants, from stillborn to one year of age, also showed that intimal thickening increased with the infants' age (Schornagel, 1956). Grading the lesions I (endothelium on regular or split elastica interna) to III (thick intima), about 40% of the males had grades II and III lesions at less than one day to one month, and 24% and 37% of the females at less than one day and up to one month, respectively. Infant boys and girls of one month to one year had grades II and III coronary lesions in 91.3% and 87.5% respectively. That the earliest coronary lesions in the youngest infants is elastica degeneration, often without overlying intimal thickening, was attested to by Levene (1956), Gillman (1959), and Kaunitz (1961). The intimal hyperplasia, usually in the areas with elastica damage, was pointed out in the early studies of Dock (1946) and Fangman and Hellwig (1947), both of whom stressed the preponderance of intimal thickening in male neonates. Because these neonatal coronary lesions are so common, there is controversy as to whether they are the earliest arteriosclerotic lesions or merely adaptive phenomena (Review: Neufeld and Vlodaver, 1971). This group confirmed the greater degree of elastica degeneration and overlying intimal fibroblastic proliferation, as well as muscle degeneration in the media, in male than in female Jewish neonates of European derivation (Ashkenazim) but found far less sex difference in intimal thickening among Yemenite (Mideastern Jewish) and Bedouin infants (Fig. 6-1) (Neufeld and Vlodaver, 1968, 1971). Histologic examination of right and left coronaries from 211 consecutive hearts from fetuses, infants, and children up to ten years of age showed significantly higher intima/musculoelastica ratios among the Ashkenazi males than among Yemenite or Bedouin males (Neufeld and Vlodaver, 1968; Vlodaver et al. 1969). Since the infants with the greatest degree of intimal damage (Fig. 6-2) were from the ethnic group with the highest rate of adult ischemic heart disease, it was considered likely that the early coronary lesions were indeed the precursors of the later coronary atherosclerotic lesions (Neufeld and Vlodaver, 1971; Neufeld, 1974).
Although coronary and myocardial lesions were most often the causative factors in the terminal event, most of the babies with coronary disease also had arteriosclerosis of other arteries, generally (in order of frequency) of the kidneys, adrenal glands, pancreas, spleen, lung, mesentery, and thyroid (Review: Moran and Becker, 1959).
6.2. Factors Suggesting Magnesium Deficiency in Infantile Cardiovascular Disease
The type of coronary arteriosclerosis, particularly of the small-to-medium coronary arteries, and the perivascular focal myocardial necrosis (that are seen in infancy) strongly resemble the coronary and myocardial lesions produced in animals on magnesium-deficient diets (Seelig and Haddy, 1976/1980). Most of the magnesium-deficient animals with cardiovascular lesions were immature. There have been comparable changes reported in herbivores however, usually during early lactation, occurring in herds grazing on magnesium-poor lands, on pasturage with factors interfering with availability of magnesium, and not infrequently in herds with a high incidence of eclampsia (Arnold and Fincham, 1950; Lynd et al., 1965; Willers et al., 1965; review: Seelig and Bunce, 1972). Data on abnormalities during pregnancy or delivery are not frequently given in papers on infantile cardiovascular disease. Some information was given in a third of the tabulated infants who died under one month of age and in less than a third of those with the disease reported in infants from one month to two and a half years of age (Appendix Table A.6A). Abnormal or frequent pregnancies, long or complicated deliveries, immaturity during gestation, multiple births, and maternal diabetes mellitus-all conditions that have been associated with low levels of magnesium-and placental insufficiency or premature separation of the placenta are conditions associated with prenatal hypoxia and malnutrition. Several of these factors were cited in eight of 29 infants dying with myocardial lesions, who had fetal distress recorded (Oppenheimer and Esterly, 1967). Such factors plus hydramnios or RH incompatibility were reported in 63 of the 157 of those under one month of age (Appendix Table A-5A) [and (Appendix Table A-5A continued)], but in only 38 of the 251 infants over one month to two and a half years old (Appendix Table A-6A) [and (Appendix Table A-6A continued), (Appendix Table A-6A continued (2), (Appendix Table A-6A continued (3), (Appendix Table A-6A continued (4), (Appendix Table A-6A continued (5), and (Appendix Table A-6A continued (6), few of whom had maternal histories cited. There were several instances in which there had been previous unsuccessful pregnancies, or in which siblings or close relatives had died similarly. Thus, it seems that metabolic disorders or gestational stress (especially in instances of maternal immaturity, or frequent or multiple pregnancies) might have played roles in absolute or conditioned magnesium deficiency. Unfortunately, magnesium levels were almost never recorded in the propositus or mother, leaving speculative the supposition that magnesium deficiency might have been contributory in the cited cases. An exception is the infant, reported by Vainsel et al. (1970), who had hypomagnesemic hypocalcemia and whose refractoriness to vitamin D and calcium therapy appeared to be familial. He and three male siblings (out of six) had had convulsive seizures. One died at six weeks; the described infant died at three months and was found to have focal myocardial necrosis and coronary calcinosis. Since he was the ninth infant in his family, both a metabolic and multiparity-induced hypomagnesemia might have participated in his severe hypomagnesemia (0.4-0.65 mEq/liter), the magnesium deficiency having been detected only a few days before death (Vainsel et al., 1970). Until prospective and retrospective magnesium data are obtained from affected infants and their mothers, from subsequent pregnancies and infants, and from near relatives, the validity of the premise that magnesium deficiency is contributory to infantile arteriosclerosis and its complications remains untested.
The medial necrosis of the coronaries seen in large infants with birth asphyxia (Gruenwald, 1949) might also be related to loss of tissue magnesium. Perhaps sufficient magnesium can leave the tissues of the coronary arteries and the heart to cause necrosis or arrhythmia or both. The intimal and medial loss of functional myocardial magnesium (Review: Seelig, 1972) might participate in the cardiac lesions of infantile cardiovascular disease.
Perhaps contributing to infantile coronary arterial lesions and microfocal myocardial necrosis (that either results in immediate death or sets the stage for cardiac death in the later months or years) is neonatal hypoparathyroidism. Lehr and his colleagues (Lehr, 1965, 1966; Lehr et al. 1966) have shown that parathyroidectomized rats, particularly when they are phosphate loaded, develop lesions of the small coronary arteries and perivascular microfocal myocardial necrosis. The high phosphate content of cows' milk, fed to infants during the neonatal period, especially when their parathyroid hormone secretion is often subnormal, and the hyperplasia of infants' coronaries might also be related to episodes of hypoxia, conceivably such as are experienced by infants that suffer from periods of sleep apnea (as in the SIDS). The production of severe arteriosclerosis, predominantly of the arterial connective tissues, by exposure of rabbits to short periods of hypoxia daily for two weeks (Helm et al., 1969; Garbarsch et al., 1969) would seem to support that supposition. Not noted in Gruenwald's (1949) study of large infants whose medial necrosis was attributed to perinatal anoxia was whether any had been born to diabetic mothers. Infants of diabetic mothers not only tend to be large but have also been found to have a high incidence of hypomagnesemia.
In the infants past the neonatal period, the use of cows' milk formulas that not only provide a substantial phosphate load, but that also provide vitamin D additional to that generally prescribed by the physician, can also contribute to magnesium deficiency. The generalized arteriosclerosis, valvular disease, and fibroelastosis of babies that have received excessive vitamin D or that are hyperreactive to it have been reviewed (Seelig, 1969b, 1978; Seelig and Haddy, 1976/1980; Seelig and Mazlen, 1977). Regarding its effects during pregnancy, experimental hypervitaminosis D has been implicated in placental abnormalities, such as those that contribute to fetal malnutrition, anoxia, and possibly to eclampsia. It is well to remember, thus, that vitamin D excess causes magnesium loss that might well be implicated in infantile cardiovascular disease (Seelig and Haddy, 1976/1980).
6.2.1. Experimental Arteriosclerosis of Magnesium Deficiency
6.2.1.1. Arterial Damage Caused by "Pure" Magnesium Deficiency
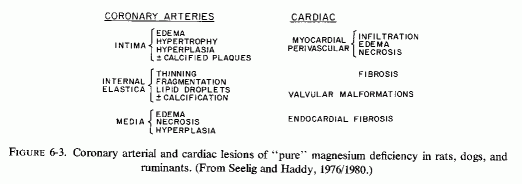
Since most of the studies of the pathogenesis of atherosclerosis have focused on fat, and most studies of magnesium deficiency were with animals (usually rats) whose control and experimental diets were also high in calcium, phosphate, and often vitamin D (Reviews: Larvor and Durlach, 197 lb; Seelig and Haddy, 1976/ 1980), there are few experimental studies of the vascular changes caused by magnesium deficiency alone (Fig. 6-3). Lowenhaupt et al. (1950) reported that young rats kept on a normal diet, except for magnesium deficiency, developed myocardial lesions (within two weeks) around the small coronary radicals of precapillary and capillary size. Other magnesium-deficiency studies, that elicited focal myocardial infiltration, necrosis, and scarring (Mishra, 1960a; Mishra and Herman, 1960; Seta et al., 1965) are suggestive of damage to the small intramyocardial coronaries. Heggtveit (1965c) reported edema of the small coronary arteries in Mg-deficient rats. Hungerford and Bernick (1976/1980) have provided details of the nature of the coronary arterial damage produced by magnesium deficiency in rats: intimal thickening with extracellular edema, thinning of the internal elastic membrane with disruption, and disorientation and hyperplasia of medial muscle cells. Some of the arteries had densely aggregated pyknotic cells in their enlarged tunica media, with narrowed lumina. This group confirmed the inflammatory changes of the perivascular myocardium, reported more than 25 years earlier by Lowenhaupt et al. (1950). Dogs on otherwise balanced magnesium-deficient diets had pyknotic intimal cells in small coronary arteries and arterioles, but no intimal hyperplasia; their medial muscle cells were loosely arranged, suggestive of edema, with necrosis and inflammation. The larger coronaries were less damaged (Wener et al., 1964). Intimal and medial calcification were described in magnesium-deficient dogs, despite decreased serum calcium levels (Syllm-Rapoport and Strassburger, 1958; Unglaub et al., 1959; Bunce et al., 1962b; Featherston et al. 1963., Morris et al., 1963; Wener et al., 1964). As in the magnesium-deficient rats, which had perivascular myocardial necrosis and edema (supra vide), magnesium-deficient dogs had comparable myocardial lesions (Unglaub et al.,1959; Wener et al., 1964). Large myocardial infarcts were seen in severely magnesium-deficient dogs (Morris et al. 1963).
The histologic arterial changes of magnesium deficiency were first characterized in cows, on spring forage on magnesium-poor soil, or where other factors interfered with the availability of magnesium (Arnold and Fincham, 1950). These observations were reaffirmed in controlled studies (Lynd et al. 1965; Willers et al. 1965). The coronary arteries and the endocardium showed intimal thickening and subendothelial degeneration and calcification of elastica fibers. Medical calcification and calcific intimal plaques were also described, as were valvular malformations and myocardial infarcts (Willers et al., 1965). The syndrome, leading to these cardiovascular lesions, was seen predominantly in lactating cows in areas where "grass tetany" or convulsions of magnesium deficiency (characterized by hypomagnesemia and hypocalcemia) occurred during late pregnancy or during lactation. Not only cows but ewes are susceptible to this disorder, and it has been noted that it is more prevalent in herds with a high incidence of toxemia of pregnancy (Herd, 1966a,b).
6.2.1.2. Arterial Damage of Magnesium Deficiency, Intensified by High Calcium and Vitamin D Intakes (Fig. 6-4)
High dietary calcium/magnesium dietary ratios have uniformly increased the susceptibility to the symptoms and signs of magnesium deficiency. It is important to remember that high intakes of calcium interfere with magnesium intestinal absorption and increase its renal excretion, and that high intakes of vitamin D also favor calcium retention over that of magnesium (Reviews: Seelig, 1964, 1971).
Cardiovascular changes, similar to those seen in "pure" magnesium deficiency, developed in rats fed 400-650 times as much calcium as magnesium (versus 40/1 in controls, which is also a much higher than normal Ca ratio). There were small inflammatory and necrotic myocardial lesions (suggestive of disease of the small coronary arteries) with increased tissue calcium and sodium, but no significant change in serum calcium, and low tissue and serum magnesium and potassium (Mishra, 1960a; Ko et al., 1962). Adding sufficient magnesium to lower the Ca/Mg ratio to 3/1 prevented the lesions (Mishra, 1960a). In a study designed to show how much magnesium is necessary to prevent macroscopically manifest intimal calcific plaques in dogs on Ca/Mg intakes of 33-50/1, Bunce et al. (1962a) found a little more than a twofold increase in grossly visible aortic intimal lesions in dogs receiving 0.6% of calcium (Ca/Mg = 33/1). Most of the dogs on high calcium/low magnesium intakes had intimal plaques.
Since vitamin D normally increases serum calcium levels and increases magnesium requirements, it is of interest that magnesium-deficient dogs on normal calcium intakes showed minimal coronary arterial calcification unless they were given vitamin D or an intravenous calcium load (Syllm-Rapoport and Strassburger, 1968; Unglaub et al., 1959). An early study (Handovsky and Goormaghtigh, 1935) showed that moderately high doses of vitamin D significantly raised the blood pressure in dogs; that vitamin D excess causes arteriosclerosis has been known even longer (Kreitmair and Moll, 1928). Like the arterial lesions of magnesium deficiency, those of experimental hypervitaminosis D (Gillman and Gilbert, 1956) involve medial and elastica degeneration and calcification (Review: Seelig, 1969), but the predominant lesions described were of the larger arteries, rather than of the coronaries. Rats on toxic doses of vitamin D also developed hypercholesterolemia, hypertension, and aortic calcification; the latter changes were prevented by high-dosage magnesium supplementation (Sos et al., 1960; Rigo, 1965; Rigo et al., 1965a; Sos, 1965).
When calves were fed low-magnesium diets that were usually comprised of whole milk or a comparable synthetic diet (both supplemented with vitamin D) for prolonged periods, they developed neuromuscular signs of magnesium deficiency and endocardial and intimal plaques, and fragmentation, degeneration, and calcification of the elastic fibers of both endocardium and arteries, and phlebothrombosis and focal myocardial necrosis (Moore et al., 1936, 1938; Blaxter et al., 1954). The magnesium-deficiency syndrome was prevented by magnesium supplementation: 30-40 mg/kg/day (Huffman et al., 1930; Duncan et al., 1935; Moore et al., 1936, 1938; Blaxter et al., 1954). Like human infants, despite these calves' high calcium intakes, their serum calcium remained normal or slightly low. In addition to the endocardial and intimal calcification, calves on a whole-milk diet developed high serum cholesterol levels (J. W. Thomas, 1959).
6.2.1.3. Arterial Damage of Magnesium Deficiency Intensified by High Fat Intakes (Table 6-1)
Studies of the influence of magnesium deficiency and its repletion on the development of atherosclerosis in rats (fed different combinations of saturated or unsaturated fat diets with and without added cholesterol and cholic acid and calcium) (pages 148-152) have shown dissociation between serum and cardiovascular lipids. It is noteworthy that increased magnesium intakes of animals on atherogenic, hyperlipidemic diets decreased arterial and myocardial lipid deposition without lowering the elevated serum lipids; the magnesium even raised the serum lipids somewhat (Vitale et al., 1957d; 1959; Nakamura et al., 1960). In contrast, high calcium intakes lowered the serum lipids but raised the arterial lipids (Vitale et al., 1957c; 1959; Hellerstein et al., 1957, 1960; Nakamura et al. 1960). Long-term administration of magnesium to rats on atherogenic diets, which only gradually lowered serum lipids to a minor degree, resulted in more rapid and significantly reduced arterial lipid deposition (Nakamura et al., 1960, 1966). The rats on the low-magnesium, high-fat diets were the only high-fat-fed rats to develop fat deposition in heart valves and plaque formation in the aorta (Nakamura et al., 1966). In this series of experiments, subintimal and medial degeneration and calcification of the elastica, as well as intimal atheromata, developed only in the rats that were magnesium deficient as well as fat loaded. Calcification of the media of pulmonary artery and of the myocardium (some with interstitial inflammatory infiltration) were also noted in magnesium-deficient, fat-loaded rats. In a further study to explore the mechanism of the intensification of atheroma formation by magnesium deficiency of rabbits on an atherogenic diet, Hirano (1966) measured the uptake of radioisotope "C-tagged cholesterol by the heart, aorta, and other viscera. Rabbits fed the magnesium-deficient atherogenic diet showed increased radioactivity in the aorta, as compared with controls. Even magnesium-deficient rabbits on low-cholesterol intakes had increased fat deposition in the aortas, but to a lesser degree. Despite the increase in aorta cholesterol in magnesium-deficient rabbits, the serum cholesterol level was not significantly altered.
Nakamura et al. (1965) found that rabbits that had developed atherosclerosis required substantial amounts of magnesium added to their diets to exert a notable effect on atherogenesis. More than 950 mg/100 g of diet was necessary to affect serum and tissue lipids. The authors commented that aortic lipid deposition is significantly enhanced by magnesium deficiency; high magnesium intake merely slows the process. The elevated intimal plaques, fragmented and calcified elastica, and mural thrombi that were reported in the magnesium-deficient rabbits, were not seen in the matched cholesterol-loaded, magnesium-supplemented rabbits; they showed no calcified lesions and less foam cells in the subintimal layer of the aorta (Nakamura et al., 1965). Narrowing of coronary arteries was noted in all of the cholesterol-loaded rabbits, but to a somewhat lesser degree in the rabbits on high magnesium intakes. Greater involvement of the small coronary arteries is suggested by the microscopic foci of myocardial necrosis in half the rabbits on magnesium-deficient, high-cholesterol diets, but in none of those that were magnesium supplemented. Bunce et al. (1962a) showed that increasing the magnesium intake, sufficiently to prevent intimal lesions in dogs on a high saturated fat diet, actually increased their serum cholesterol levels, whereas the dogs on the highest Ca/Mg ratio had lower serum cholesterol (270%) and more intimal lesions. Dogs on a magnesium-free, corn-oil-rich, low-calcium diet had intimal thickening and plaques with narrowed coronary lumens, but minimal lipid deposition (Vitale et al., 1961). Monkeys on a similar diet exhibited raised intimal atheromata and fibroblastic intimal thickening, with disrupted elastica, but no arterial calcification.
These findings raise the question as to whether seeking to correlate serum magnesium and cholesterol levels provides meaningful data regarding the influence of magnesium deficiency or therapy on atherosclerosis. Even when high doses of magnesium are given to hypercholesterolemic animals, the changes in serum lipids are less consistent than is the lowering of tissue lipids. The serum levels of cholesterol have been unaffected, or even raised in some of the studies; the β-lipoprotein fraction seems to be influenced somewhat more. Although magnesium deficit intensifies atheromatosis, it takes quite large doses and/or prolonged administration of magnesium to protect against the disease in hyperlipemic animals (Hellerstein et al., 1957; Rigo et al., 1963, 1965a,b; Nakamura et al., 1965, 1966). It seems that neither the serum magnesium nor cholesterol level are illustrative of the tissue levels. Thus, to determine the effect of magnesium on lipids in man, we must investigate the response to effective doses of magnesium. The preliminary clinical trials cited in this chapter are not conclusive. Prolonged trials with more intensive exploration of the leads mentioned here are indicated. The effect of magnesium on high-density lipids needs study.
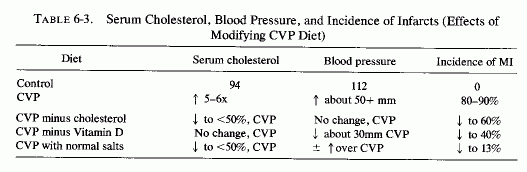
6.2.1.4. The Cardiovasopathic (CVP) Diet
An experimental diet (Table 6-2) has been devised that causes spontaneous myocardial infarctions (MI) in 80-90% of the animals (rats, dogs, and cocks) fed that diet but kept under otherwise normal conditions (Sos et al., 1960, 1964a,b,c; Sos, 1965; Rigo et al.,1961, 1963a,b, 1965a,b; Rigo, 1971; Gati et al., 1964, 1965; Szelenyi, 1971, 1973; and Review: Seelig and Haddy, 1976/1980). With the exception of low chloride, it possesses the characteristics of diets consumed by many in our affluent society. It is high in fat, cholesterol, vitamin D, sodium, phosphate, and protein; it is low in magnesium, potassium, and chloride. In addition to the massive infarctions, animals on the CVP diet had atherosclerosis, hyperlipidemia, and the abnormalities (Table 6-3). Without the added cholesterol, animals on the (modified) CVP diet still had high cholesterol levels, but they were half as high as those on the complete CVP diet. The hypertension was unaffected, but the incidence of MI dropped to 60% of the group. Elimination of only vitamin D did not lower the blood cholesterol, but the animals had only slight hypertension, and fewer (40%) developed MI. Halving the protein content of the CVP diet (to a normal intake) resulted in a slight increase in serum cholesterol, no change in the hypertensive level, but resulted in about half the MI incidence (40%) of the CVP animals. Providing a normal salt mixture lowered the cholesterol somewhat but not the hypertension. It lowered the incidence of infarction to 13%. Increasing the dietary intake of magnesium chloride fivefold over the normal requirement mitigated, significantly, the cardiopathic changes as well as the coronary and aortic pathology, which had included thickening of the small coronary arteries, with marked increase of the arterial wall/lumen ratio (Sos, 1965; Szelenyi, 1971). The increased magnesium intake also reduced the extent of damage produced by such intensifying factors (added to the CVP diet) as neurogenic stress, or ACTH. When the CVP diet was modified by increasing the cholesterol threefold, the fat fourfold, vitamin D and cod liver oil 1/3 each, and adding thiouracil, marked hypercoagulability was produced. Fivefold increased magnesium intake restored the coagulation and prothrombin times to normal (Szelenyi, 1971, 1973).
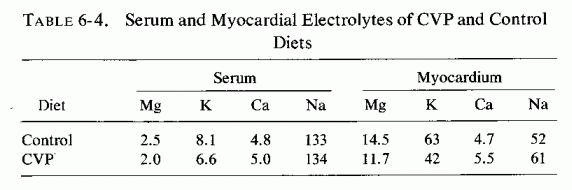
The rats on the CVP diet retained 15 times as much sodium as did the controls, but their myocardial and serum sodium levels differed little from control values. Their myocardial calcium rose 12%, but their serum calcium remained essentially unchanged. Their myocardial magnesium and potassium levels dropped 19 and 33%, respectively; serum values of both cations dropped about 20% (Sos, 1965; Table 6-4).
6.2.1.5. Other Cardiovasopathic Models That Might Entail Relative Magnesium Deficiency
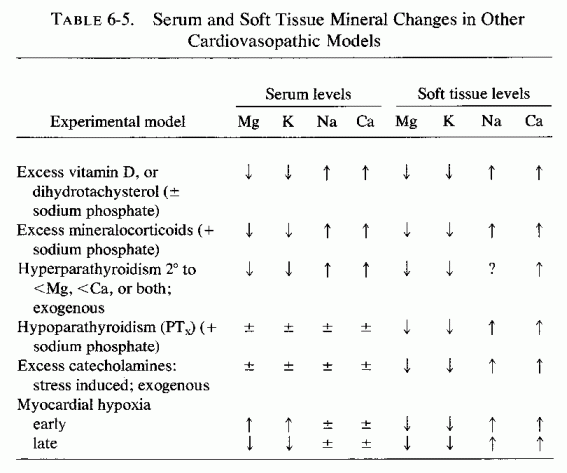
Arterial lesions, similar to those produced by magnesium deficiency, in combination with high calcium, vitamin D, or fat intakes or other imbalances (i.e., CVP diet) have been produced by modalities that increase serum calcium or cholesterol levels, increase tissue sodium and calcium levels, and decrease magnesium and potassium, both in the serum and in the tissues. (Table 6-5). Dihydrotachysterol, particularly in combination with sodium acid phosphate (NaH2PO4) causes coronary and aortic calcification and periarteritis, lesions that are intensified by magnesium or potassium deficiency, partially protected against by administration of either cation and better protected against by both and by the chloride ion. (Selye, 1958a,b; Bajusz and Selye, 1959; Mishra, 1960d). Mineralocorticoids plus phosphates produce multifocal necrosis (suggestive of small coronary disease), the intensity of which is also increased by magnesium and/or potassium deficiency; again, each cation is protective (Selye, 1958a,d,e,f; Selye and Mishra, 1958; Bajusz and Selye, 1959; Mishra, 1960b; Selye and Gabbiani, 1965). Parathyroid extract, with sodium phosphate salts (Selye, 1958c; Lehr, 1963) or stimulation of parathyroid secretion and/or adrenal medullary and cortical secretion, as occurs in renal damage or nephrectomy (Lehr, 1959), causes subintimal arterial damage with calcification of the damaged elastica, in addition to myocardial infiltration and edema. Administration of mineralocorticosteroids markedly intensifies the cardiovascular lesions of these (Lehr, 1959) and of the catecholamine myocardial necrosis model (Guideri et al., 1971). Paradoxically, despite the calcium-mobilizing effect of parathyroid hormone, and the vitamin-D-like arterial damage it produces in combination with a phosphate salt, Lehr (1959) has shown that phosphate-loading of parathyroidectomized rats causes even more severe cardiovascular lesions. Subsequent work from his laboratories has demonstrated that the common denominator in the experimental models-calcium- or phosphate-loading in the presence or absence of parathyroid hormone, or with mineralocorticoid, or catecholamine (exogenous or endogenous)-is depletion of myocardial magnesium and subsequently of potassium (Lehr, 1965b, 1969; Lehr et al., 1966, 1969, 1970/1972, 1976/1980). The increase of cellular calcium reflects, predominantly, the calcification of injured tissues, even in the presence of hypocalcemia of the parathyroidectomized rats. Stress has also been associated with markedly increased myocardial damage when the animals are magnesium or potassium deficient, and magnesium administration has protected against stress and exogenous catecholamine-induced cardiovascular damage (Selye, 1958g; Selye and Mishra, 1958; Shimamoto et al., 1959; Mishra, 1960e; Mishra and Herman, 1960; Bajusz, 1965a). Lehr (1965, 1966) has correlated the microfocal myocardial necrosis, seen in most of the drug- and stress-related experimental cardiovasopathic models (which resemble the lesions of "pure" magnesium deficiency, supra vide), with damage to the cardiac microcirculation, with medial degeneration and perivascular myocardial necrosis, and has stressed the depletion of intracellular magnesium as an early and consistent change. The animals that are loaded with calcium, vitamin D, and/or fat: all agents that cause hypercholesterolemia, hypertension, or thrombogenesis seem to have a greater tendency to develop infarcts (supra vide: CVP diet). That magnesium deficiency predisposes to the hypercoagulability, and that magnesium administration has been protective, may relate to the role of magnesium in platelet function (Review: Elin, 1976/1980), as well as to the effects of magnesium on coagulation factors (Szelenyi et al., 1967; Szelenyi, 1971, 1973; Stevenson and Yoder, 1972; Seelig and Heggtveit, 1974).
6.3. Catecholamine-Induced Arterial Damage; Magnesium Interrelationships
In addition to the increased susceptibility to atherogenesis that catecholamines can cause by inducing lipolysis, Raab (1958) called attention to the evidence that prolonged administration of small doses of epinephrine produces intimal thickening of small and large vessels of rabbits and dogs, and that larger doses produce necrotizing and calcifying lesions of the media. The similarity of these arterial lesions to those of magnesium deficiency, particularly in association with high intakes of calcium or of calcemic agents (supra vide), brings attention to the evidence that catecholamines cause loss of cellular magnesium. Epinephrine has been shown to increase plasma magnesium levels after its injection or after drug- or stress-induced stimulation of its secretion (Rogers and Mahan, 1959a,b; Larvor, 1968; Larvor and Rayssiguier, 1971; Rayssiguier and Larvor, 1971/1973). Catecholamine injection or its stress-induced secretion has caused lowered myocardial magnesium levels. This effect might be partially reciprocal to catecholamine-induced cellular uptake of calcium, a physiologic action that contributes to its positive inotropic effect (Nayler, 1967). The arterial damage caused by catecholamines, however, must be a pathologic extension of its activity that intensifies production of a low cellular magnesium/calcium ratio.
One mechanism might be via local hypoxia mediated by proliferative constrictive endothelial proliferation, in conjunction with its increase of oxygen consumption (Raab, 1969). It should be recalled that even short-term local hypoxia, such as is produced by occluding the vessels by a blood pressure cuff can cause increased plasma magnesium, presumably as a result of egress of cellular magnesium (Whang and Wagner, 1966; S. P. Nielsen, 1969). Thus, this mechanism, too, can produce a low Mg cellular ratio.
The general increase in blood pressure that is the classic response to catecholamine release or injection must also be considered. The cardiac output increases as a result of increased strength of myocardial contraction and increased heart rate [both contributed to by the catecholamine-stimulated shift of calcium into the heart (Nayler, 1967)], and secondary to the increased venous return to the heart, as splanchnic, renal, skin, and mucosal arterioles constrict.
The protection by magnesium against intimal damage (supra vide) might serve to protect the arterial lining from the mechanical stresses caused by sudden changes in pressure and local oxygenation. There are experimental data suggesting that magnesium deficiency increases some of the catecholamine effects on the arteries, and that magnesium excess tends to counteract them. For example, Hanenson (1963) found that absence of magnesium from the medium in which aortic slices were suspended markedly increased the contractile response to norepinephrine; its addition decreased the contractile response. However, recent work on interrelationships of magnesium and calcium with vasoactive hormones on vascular muscle has elucidated the magnesium dependence of the reactions and explained how magnesium depletion can cause refractoriness to vasoactive hormones (pages 179-183). In vivo rat studies have shown that slow intravenous infusion of magnesium sulfate decreases the hypertensive response to epinephrine or norepinephrine (Cession et al., 1963). The influence of magnesium deficiency or excess on release of catecholamines is considered under our discussion of magnesium and the heart. In this regard, the demonstration that arterial tissue exhibits rapid uptake of catecholamines (particularly of epinephrine) even when injected within the range that is probably produced by catecholamine-releasing agents, such as nicotine, stress, hypoglycemia, or thyroid hormone (Raab and Gigee, 1958) is probably relevant to the clinical situation.
A high-fat diet that was thrombogenic (incorporating propylthiouracil and cottonseed oil) became cardiopathic when NaH2PO4) was added (Savoie, 1972a,b, 1975). Since the findings with this regimen, and with modifications of the electrolyte steroid cardiac necrosis (ESCN) syndrome developed by Selye (supra vide), including catecholamines and exposure to stress, point predominantly to effects of drugs and minerals (predominantly magnesium) on cardiac metabolism (Savoie, 1971a,b, 1975) infra vide.
6.4. Magnesium Deficiency, Mast Cells, and Arteriosclerosis
Still another change has been seen in the cardiovascular tissues of both atherosclerotic experimental animals and patients, and in experimental magnesium deficiency: decreased numbers of mast cells and evidence of degranulation. It has been suggested that connective tissue mast cells may play a role in the development of arteriosclerosis (Constantinides, 1953; Cairns and Constantinides, 1954; Wexler (1964). Evidence for this theory derives from the observation that rats, a species resistant to atherosclerosis, has many mast cells in the myocardium, whereas susceptible rabbits (Constantinides, 1953) and chickens (Padawer, 1957) have few mast cells. Furthermore, young women have more mast cells than do young men and atherosclerotic patients have fewer mast cells than do normals (Hellstrom and Holmgren, 1950; Constantinides and Cairns, 1954). Wexler (1964) has reported that the number of myocardial mast cells was most severely depressed in breeder rats that developed the most severe spontaneous arteriosclerosis, and that mast cells were not found in the vicinity of the arteriosclerotic lesions. Their granules showed a marked change from metachromasia to orthochromasia, which was interpreted as indicating secretory discharge. The decrease in numbers of mast cells in arteriosclerotic arteries has been correlated with the decrease in hyaluronic acid in arteriosclerotic arteries (K. Meyer et al., 1959; Kaplan and Meyer, 1960; Buddecke, 1962). Since mast cells secrete hyaluronic acid (Padawer, 1957), the decrease in number of mast cells in arteriosclerosis may explain the decline in hyaluronic acid content. Experimental magnesium deficiency has also been shown to cause decreased tissue mast cells, and to increase their degranulation (Belanger et al., 1957, Bois et al., 1960; Hungerford, 1964; Hungerford and Bernick, 1976/1980). More data are required to ascertain whether the decreased numbers of mast cells in animals and patients with atherosclerosis might be contributed to by magnesium deficiency.
6.5. Arterial Resistance, Blood Pressure, and Magnesium
The term "hypertensive-arteriosclerotic cardiovascular disease" reflects the frequent association of what are actually two separate diseases: hypertension and arterio-, or more commonly, atherosclerosis. That hypertension can lead to arterial damage has been accepted for many years, although the mechanisms are still subject to dispute. This is not the place to consider the experimental evidence and hypotheses that suggest that hypertension can predispose to formation of atherosclerotic by damaging the endothelium and permitting diffusion of cholesterol into the intima (Haust, 1970), e.g., at sites of swirling and eddying of the blood stream (Duff, 1951), as a result of pressure-induced arterial dilatation (Schornagel, 1956; Helm et al., 1971). It is, however, pertinent that each disorder has been found in magnesium-deficient models, most commonly in combination.
The mechanism of the morphological changes produced in the blood vessels by magnesium deficiency is not clear, but the changes almost certainly contribute to the increased arterial resistance. A contribution by vasoconstriction also seems likely, particularly since magnesium deficiency (experimental) is usually associated with decreased serum concentrations of magnesium, potassium, and in soft tissues, decreased content of magnesium and potassium and increased content of calcium and sodium. Depending on whether the magnesium deficiency is associated with intake of calcemic agents, the serum calcium can be either low, normal, or high. When it is high, the electrolyte imbalance is one that has been shown to increase arterial resistance (infra vide). Increased plasma renin activity, blood serotonin level, and urinary aldosterone excretion have also been noted in magnesium deficiency-all factors that also increase arterial resistance.
The effect of magnesium deficiency on blood pressure involves complex interactions. Although most of the experimental models are associated with increased blood pressure, there are both clinical and experimental circumstances in which no effect, or actual lowering of blood pressure has been seen with magnesium deficiency. Insight into these paradoxical findings derives both from in vivo magnesium-deficiency studies and from in vitro investigations that have elucidated several aspects of the response of the vascular smooth muscle contractility and resistance to changes in magnesium and calcium concentrations.
6.5.1. Increased Arterial Resistance: Low Mg + K, High Ca + Na
Among the experimental models of cardiovascular disease are several that are characterized by decreased magnesium levels, and that are associated with increased arterial resistance. The magnesium-deficient animals that develop hypertension, as well as arterial and cardiac damage, usually also have hyperlipidemia, atheromata, and hypercalcemia. Animals with vitamin D toxicity, or those given the CVP diet (supra vide), fall into this category. Only in one study, in which the relative dietary intakes of magnesium and calcium were changed, did the blood pressure of calcium-deficient, magnesium-adequate rats show a greater decline than did that of magnesium-deficient rats (Itokawa et al., 1974b).
Acute studies, in which a low ratio of magnesium, potassium, or both, to calcium is produced by intra-arterial infusions, have shown that such a ratio (particularly in the presence of alkalosis) increases arterial resistance in peripheral, renal, and coronary circulation (Haddy, 1960, 1962; Haddy et al., 1963, 1969; J. Scott et al., 1961, 1968; Frohlich et al., 1962). On the basis of the experimental findings, Haddy and Overbeck (1962) and Frohlich et al. (1964) suggested that such electrolyte imbalances might be a common denominator in clinical hypertension. They cited hypercorticism, hyperparathyroidism, vitamin D toxicity, and eclampsia as diseases characterized by hypertension and often by such electrolyte abnormalities. Similar acute intraarterial infusion studies with normotensive and hypertensive human subjects yielded comparable results (Overbeck et al., 1969). The most recent reviews consider the physicochemical interrelationships among the cations and the arterial contractile processes (Altura and Altura, 1977a,b; Haddy and Seelig, 1976/1980). Calcium plays a central role in excitation-contraction coupling and this ion competes with magnesium for binding sites on the membrane of the vascular smooth muscle cell. When extracellular magnesium falls, more calcium is available on the membrane for entrance into the cell with each spike potential. Furthermore, the number of spike potentials may increase because lack of magnesium and potassium suppresses the sodium-potassium pump, which, because of the electrogenic nature of the pump, leads to a decrease in the resting membrane potential. Increased membrane calcium and number of spikes would elevate intracellular calcium and cause vasoconstriction.
The magnesium concentration of the medium in which arterial strips are suspended affects their contractile response to vasoactive drugs and hormones. In its absence, potent contractile responses have been produced (Altura, 1970, 1975a,b; Altura and Altura, 1971, 1974, 1976/1980, 1978; Altura et al., 1976/1980), an action that might be attributable to the greater surface binding of calcium when there is no competition by magnesium for common binding sites (Altura and Altura, 1971, 1974; Turlapaty and Carrier, 1973; Jurevics and Carrier, 1973). Somlyo et al. (1972) have proposed that incubation of arterial strips in magnesium-free solutions reversibly blocks the hyperpolarizing effect of cyclic AMP. Altura and Altura (1977, 1978) have hypothesized that these acute effects of changes in extracellular ionic magnesium result from effects on calcium permeability, translocation, and membrane stability, as well as from competition for binding sites. In fact, as with other tissues, exposure of vascular smooth muscle to low extracellular magnesium concentrations has resulted in increased tissue calcium content (Altura and Altura, 1971; Palaty, 1971, 1974). It can be speculated that reciprocal changes in serum and tissue calcium seen in magnesium-deficient animals (Review: Seelig and Haddy, 1976/1980) might be mediated by these mechanisms. Altura and Altura (1974) have provided evidence from their in vitro studies that withdrawal of ionic magnesium from the suspending medium produces arterial muscle contraction that is due to the inward movement of calcium. When a chelating agent (Ca EDTA) that selectively removes magnesium is added to the medium, the arterial muscle contracts; when one that selectively chelates calcium (EGTA) is added, the arterial muscle relaxes (Altura and Altura, 1975). Carrier et al. (1976) have shown that magnesium has two components in its interaction with calcium: (1) competition at extracellular sites, probably at the membrane; and (2) intracellular competition at sequestration sites. They confirmed the increased tension of arteries in a potassium-free medium (Hendrick and Casteels, 1974) and showed that magnesium decreases arterial sensitivity to calcium in both high- and low-potassium solutions, but increases the maximum calcium-induced response in high-potassium solution.
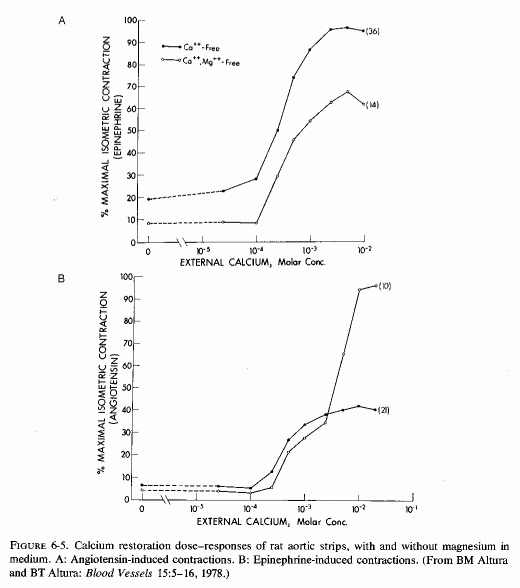
Changing magnesium and calcium concentrations affects the vasocontractile responses to hormones. At such low rates of magnesium infusion in dogs as to barely affect arterial resistance, the arterial contraction produced by injected catecholamines was substantially reduced (Haddy, 1960; Frohlich et al., 1962). Suspension of arterial strips in media lacking both calcium and magnesium resulted in almost no contractile response to such agents as acetyl choline, angiotensin, or epinephrine; restoring the calcium but not the magnesium markedly increased the vascoconstriction (Altura and Altura, 1978, 1976/1980) (Fig. 6-5)
The converse effect, that of the vasodilatory effect of high magnesium concentrations (Haddy, 1960; Haddy and Scott, 1965; Scott et al., 1968; Overbeck et al., 1969), seems to be mediated by displacement by magnesium of calcium bound to the cell surface. This has been shown to inhibit calcium influx and to uncouple excitation from contraction in myocardial cells (Langer et al., 1968; Shine and Douglas, 1974), and is probably also true for vascular muscle. Possibly, in this circumstance, the excess magnesium that displaces calcium from surface binding sites allows for fewer depolarizations and less contractility. Also to be considered is the possibility that high levels of magnesium markedly decrease the hypertensive response to angiotension II, as has been shown in rats (Cession et al., 1963).
6.5.2. Magnesium Deficiency and Decreased Blood Pressure; Refractoriness to Vasoactive Hormones
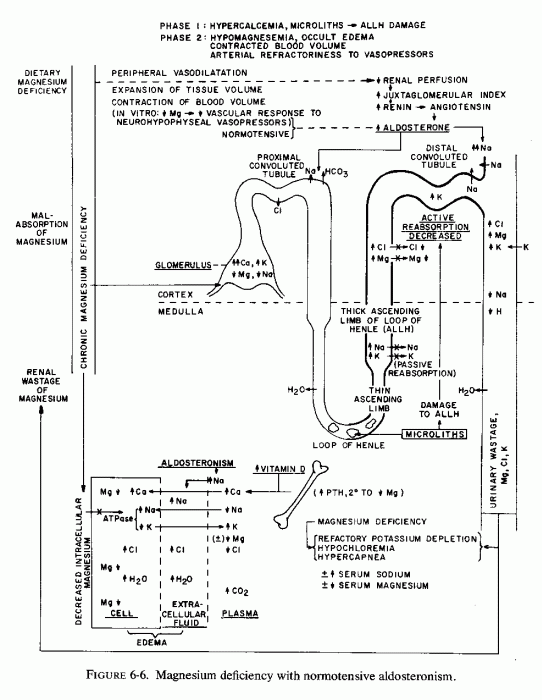
In contrast to the above observations, and in conflict with the "logical" explanation of mechanisms by which magnesium deficiency causes vasoconstriction and its excess causes vasodilation, there is both experimental and clinical evidence that magnesium deficiency has caused decreased blood pressure (Fig. 6-6). The demonstration by Cantin (1970, l971/l973a,b) that magnesium-deficient rats develop a continuous increment of the juxtaglomerular index (JGI), and of the width of the zona glomerulosa of the adrenal cortex, explains the aldosteronism of magnesium deficiency reported by Ginn (1968) but not the lack of hypertension in Cantin's deficient rats. He commented on the similarity of the JGI changes produced by magnesium deficiency to those reported after adrenalectomy, sodium deficiency, or renal ischemia (Cantin, 1971/1973a), and considered it plausible that increase in the JGI and the widening of the adrenal cortical zona glomerulosa might reflect response to a shift of fluid from the vascular space (with decreased circulating volume), as occult edema developed (Cantin, 1970, 1971/1973b). He suggested that since the rise of the JGI and the adrenal cortical changes developed early (by the 15th day) in his magnesium-deficient rats, the shifts in sodium and potassium content of serum and urine, and the subsequent marked edema (by the 25th day) might indicate stimulation of the renin-angiotensin-aldosterone system (Cantin, 1970). It was postulated that the JGI and adrenal cortical changes were probably mediated by diminution of the arterial pressure of the renal afferent arterioles, which led to stimulation of the renin-secreting, granular cells of the juxta-glomerular area, with subsequent angiotensin production and stimulation of aldosterone secretion (Cantin, 1970; Cantin and Huet, 1973). In the magnesium-deficient rats of Dagirmanjian and Goldman (1970), the systemic blood pressure was unaffected. They found the blood flow to be diminished by as much as 50% in most organs in the deficient rats that survived 40 days, but that there was splanchnic (gut and liver) vasodilatation that earlier had balanced the visceral vasoconstriction (in terms of systemic blood pressure). Early blood flow changes in these rats included increased flow to the adenylhypophyseal area.
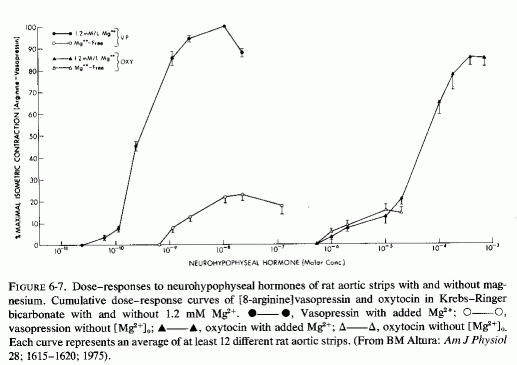
It is suggested (Haddy and Seelig, 1976/1980) that this might be a response to the decreased activity of neurohypophyseal peptides in magnesium deficiency. For example, it has been suggested that magnesium potentiates the contractile response of isolated vascular smooth muscle to vasopressin, oxytocin, and vasotocin, the action of which is magnesium dependent (Somlyo et al., 1966; Somlyo and Somlyo, 1970). Altura (1975a,b; Altura and Altura, 1977) have elucidated the mechanisms by which magnesium enhances the vasoconstrictor response to the vasoactive peptides. Thus, in the absence of optimal magnesium concentrations, the arteries exhibit refractoriness to high levels of the neurohypophyseal peptides. In fact, Altura (1975) clearly showed that the isometric contraction in response to vasopressin was markedly diminished in the absence of magnesium, as compared to its response in the presence of normal magnesium concentration (Fig. 6-7). Responsiveness to angiotensin was absent when both calcium and magnesium were missing from the medium; it was greatly increased when the calcium was restored in the absence of magnesium (Altura and Altura, 1976/1980b). This observation confirms the early in vivo observation that calcium but not magnesium is necessary for angiotension-II-induced hypertension (Cession et al.,1963).
Still another magnesium-vasoactive hormonal interrelationship has been elucidated by Altura et al. (1976, 1976/1980). They have shown that without optimal magnesium in the bath fluid suspending isolated rat arterial muscle, prostaglandin cannot evoke arterial muscle relaxation.
6.5.3. Clinical Magnesium Deficiency and Blood Pressure
With so many magnesium-related factors influencing arterial contractility, it is not easy to select those that will precipitate either hypo- or hypertension, or produce symptomatic signs of magnesium deficiency without notably affecting the blood pressure. The most dramatic changes in blood pressure mediated by magnesium are (1) the rise that has occurred during iatrogenic hypomagnesemia, produced by replacement of gastrointestinal and renal losses by magnesium-free fluids, which have been lowered by magnesium repletion (Smith et al., 1960; Smith, 1963; Hall and Joffe, 1973); and (2) the hypotension seen with severe hypermagnesemia (Mordes et al., 1975). Less frequently noted is the hypotension of severe magnesium depletion as in children with the recovery syndrome of protein calorie malnutrition (Caddell, 1965, 1967).
In general, gradual or chronic changes in serum magnesium levels are not associated with marked changes in blood pressure. On the other hand, note should be taken of the hypomagnesemic form of aldosteronism (Mader and Iseri, 1955; Mime et al. 1957) that is usually associated with moderately to markedly elevated blood pressure. In contrast, a woman has been reported who had marginal magnesium deficiency, occult edema, signs of latent tetany, and subnormal blood pressure, with intermittent aldosteronism and hyperreninism (Seelig et al. 1975, 1976/ 1979), a syndrome much like that reported by Cantin in rats (Cantin, 1970, 1971/1973).
Whether magnesium deficiency contributes to the hypertension of children with the supravalvular aortic stenosis syndrome that is associated, not only with hypercalcemia, but hyperlipidemia and that has been associated with hyperreactivity to vitamin D (Reviews: Black, 1964; Beuren et al., 1962, 1964, 1966; Seelig. l969b; Seelig and Haddy, 1976/1980) remains to be ascertained. There have been a few instances of hypomagnesemia reported, but the use of milk of magnesia to control the common constipation of this syndrome makes it difficult to interpret the rare reports of magnesium levels. Hypertension and hyperlipidemia have been reported in children and adults with hypervitaminosis D (Frost et al., 1947; Lang and Eiardt, 1957; DeLangen and Donath, 1956; Beuren et al. 1964, 1966; and 24 cases in Appendix Table VIa; low serum magnesium levels have been reported only rarely (Frost et al., 1947; Lowe et al.,. 1954). Dalderup (1960) speculated that the damage of infantile hypercalcemia might be related to cellular magnesium deficiency. Other conditions associated with hypercalcemia and hypomagnesemia in which hypertension is not uncommon include hyperparathyroidism (Pyrah et al., 1966) and hemodialysis with "softened" water (Schulten et al., 1968).
The relationship of the magnesium status to adult hypertensive syndromes is difficult to ascertain. Most of the emphasis has naturally been on sodium/potassium exchanges. The potentiation of the pressor effects of catecholamines by corticosteroids, which was demonstrated by Raab, and correlated with the transmembrane Na/K gradient and blood pressure regulation (Raab, 1959), can also be referred to as regards magnesium/calcium shifts. Both hormones cause magnesium egress from the cells; catecholamines also increase calcium influx.
As has been discussed, hypomagnesemia is common in preeclampsia and eclampsia, and hypotensive as well as anticonvulsive response to magnesium therapy in pharmacologic doses is anticipated. However, still to be proved is whether these responses reflect repair of a deficit or merely vasodilation in response to a pharmacologic agent. Similarly the use of magnesium to control hypertensive crises of renal disease (during the diuretic phase) requires resolution as to mechanism. In hypercalcemic hypertension, renal damage may complicate the diagnostic problem.
As regards essential hypertension, the common use of diuretics that cause renal magnesium loss makes interpretation of serum magnesium levels difficult Holtmeier (1969b) has surveyed cardiovascular and other complications of diuretic treatment of hypertension that might result from magnesium loss and recommends its repletion.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}