MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part II: Chapter 7
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
7
Magnesium Deficiency/Loss from Myocardium
The foregoing section has dealt predominantly with the evidence that magnesium deficiency can be contributory to arterial lesions (culminating either in sudden death or in chronic atherosclerosis) that are implicated in the cardiovascular diseases of civilization. Raab (1972), in his introduction to a symposium on myocardiology, commented that the current "official" approach to the problem of degenerative heart disease represents adherence to "traditional but outdated concepts that imply a purely, or almost purely coronary vascular origin of fatal myocardial lesions." He referred to evidence that in about half the deaths clinically attributed to "myocardial infarctions," "coronary occlusions," "coronary thrombosis," or "coronary artery disease," no thrombi or vascular occlusions were found on autopsy (Baroldi, 1969, 1970/1972; Spain and Bradess, 1960). He suggested that the term "coronary heart disease" be replaced by one referring to "cardiac hypoxic dysionism," as encompassing the ionic myocardial changes produced in association with the myocardial hypoxia resulting from a decreased oxygen supply (coronary insufficiency) in conjunction with stress-induced hormonal (catecholamine) increased oxygen demand (Raab, 1969). As depicted in Fig. 7-1 (Raab, 1972), hypoxia causes decreased myocardial concentrations of both magnesium and potassium and increased myocardial sodium. This dysionic pattern is contributed to by stress-induced corticosteroid secretion.
When myocardial levels of magnesium fall, there are many contributory factors. That nutritional imbalances leading to general magnesium deficiency, such as have been described in the introductory chapter on epidemiology, can contribute and can reduce the resistance of the myocardium to stress and to noxious agents seems likely. It might be the extra magnesium that is provided by hard water that is responsible for the much lower incidence of sudden death from ischemic heart disease (IHD) in residents of hard-water areas, as compared with the IHD sudden- death rate in soft-water areas. It is possible that water-magnesium is sufficient to correct (at least partially) the marginal magnesium deficiency that has been shown to be prevalent and increasing in the United States and in Europe. The lower myocardial and coronary magnesium levels found in accident victims in soft-water areas, as compared with those found in comparable subjects in hard-water areas, indicate that an insufficient magnesium intake is reflected by lower myocardial magnesium levels (T. Crawford and M. D. Crawford, 1967; T. Anderson et al., 1973, 1975, 1976/1980). It is important to note that despite the difference in myocardial magnesium levels, plasma magnesium levels in residents of soft- and hard-water areas are the same (T. Anderson et al., 1975, 1976/1980). This is another indication of the unreliability of plasma magnesium as an indication of the total status of magnesium or of its level in vital organs.
7.1.
Cardiac Magnesium Lability
Although, on the grounds of logic, one would expect the heart to retain magnesium with avidity-since substantial loss of myocardial magnesium is incompatible with life-cardiac magnesium is actually quite labile. Rogers and Mahan (1959 a,b) reported that in the exchange of plasma magnesium with tissues, there are rapidly and slowly exchangeable forms of magnesium in the tissues of rats. In the heart, liver, and kidney, the exchange was rapid, reaching equilibrium in about three hours. In cows and calves, the equilibration was slower than in rats, but liver, kidney, heart and pancreas similarly showed most rapid exchangeability of 28Mg (Rogers et al., 1964). Page and Polimeni (1972), also working with rat hearts, have demonstrated that about 98% of ventricular cellular magnesium is exchanged at the same but relatively slow rate [disagreeing with Rogers and Mahan (1959 a,b) that the exchangeable portion was "rapidly" exchangeable]. They found that only 2-3% is inexchangeable (Page et al., 1972), in contrast to that of skeletal muscle, 75- 80% of which is exchangeable (Gilbert, 1960). They found that the rate of myocardial magnesium exchange is 0.15 ± 0.02 mM Mg/minute/kg-1 dry ventricle or about 0.21 ± 0.02 pmol/sec/cm-2 of plasma membrane. The rate of exchange is independent of the rate of contraction or the external work done by the ventricle (Polimeni and Page, 1973a,b, 1974). Measurements of the influx and efflux of magnesium and the very low passive permeability of myocardial cells to magnesium suggested that there is probably a carrier-mediated mechanism for its cardiac transport that might be capable of preventing development of unphysiologically high myocardial cellular levels of magnesium (Page and Polimeni, 1972). Most of the 98% of exchangeable myocardial magnesium is presumably present as Mg complexes of the adenine nucleotides: ATP, ADP, and AMP. Less than 15% is associated with the mitochondria or myofibrils (Polimeni and Page, 1973a). The mitochondrial mechanism of magnesium transport has been shown in vitro to cause accumulation of large amounts of magnesium by transporting it across the inner mitochondrial membrane into the matrix (Brierley et al., 1962; Brierley, 1967; 1976). More recent studies show that there are mitochondrial ionophores that mediate magnesium (and other ions) transport (Green et al., 1975) and that such ionophores have been identified in heart mitochondria (Blondin, 1974, 1975). In rat ventricles all of the mitochondrial magnesium is exchangeable with 28Mg given intraperitoneally (Page and Polimeni, 1972), and so possibly this may be a means of regulating the amount of ionic magnesium in the cytoplasm. in vivo, myocardial cells accumulate a proportional increase in magnesium in response to stimuli that cause cellular hypertrophy (such as mechanical constriction of the ascending aorta); under such conditions there is also a disproportionate increase in sequestered myofibrillar magnesium (Page et al., 1972). Polimeni and Page (1973b, 1974) comment that a constant cellular magnesium concentration is essential to the myocardial cell. They observe that since a major proportion of cellular magnesium is complexed with the adenine nucleotides, it may be the constancy of the magnesium concentration that is related to the constancy of adenine nucleotide concentrations, which is necessary for normal myocardial cellular metabolism, including ionic exchange and energy production.
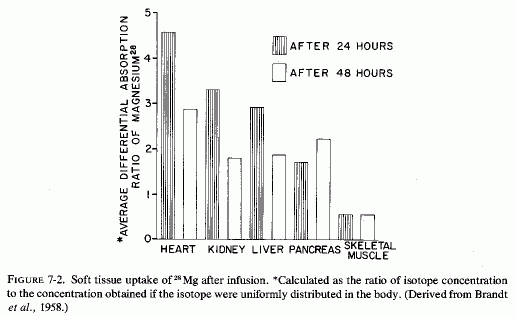
The ready exchangeability of almost all myocardial magnesium, and the demonstration of Mg2+-K+, specific mitochondrial ionophores that mediate myocardial mitochondrial magnesium transport, probably explain its rapid uptake, which is demonstrable when it is given as 28Mg Brandt et al. (1958) reported that of all the soft tissues and viscera analyzed from 24 to 48 hours after 28Mg administration (i.v. infusion to rats), the heart took up the greatest proportion of the isotope. [The kidneys, liver, and pancreas took up less, but much more than did the other tissues studied (Fig. 7-2)]. They suggested that finding such marked avidity for the magnesium, in tissues with high enzyme activity, was not surprising, in view of the importance of magnesium in ATP and other enzyme systems (Lehninger, 1950; Green and MacLennan, 1960). The heart's uptake of 28Mg has been shown to be ten times as rapid as that of skeletal muscle (Brandt et al., 1958; Aikawa et al., 1959; Field, 1961; Field and Smith, 1964). In view of their demonstration of the particular avidity of dogs' hearts for 28Mg in dogs, Glaser and Brandt (1959) extended the study to calves and rabbits. The findings were consistent in the three species. They found the greatest avidity for 28Mg in the interventricular septum and in the left ventricle of calves. The authors postulated that the high 28Mg uptake of the septum might reflect the requirement of the conduction system for impulse transmission. The greater septal and left ventricular uptake of 28Mg than in the rest of the heart was reaffirmed in dogs (Glaser and Gibbs, 1962). Lazzara et al. (1963) confirmed the avidity of ventricles and septum for tagged magnesium at 46, 56, and 68 hours after its i.v. administration in dogs. It is of interest that in an analysis of different portions of the heart (of dogs), Burch et al. (1965) found the highest cardiac concentrations of magnesium in the interventricular septum, epicardial myocardium, and ventricles. Rogers et al. (1964) did not specify the portions of the heart scanned, but included that organ as one of those with the highest specific activity 2-6 hours after injection of 28Mg into cows and calves.
In magnesium-deficient rats, the relative specific activity (the ratio of the specific activity of the tissue to that of plasma) of heart and other metabolically active organs (e.g., kidney, liver, glandular tissue) reached peak levels within 2-3 hours after injection of 28Mg, and remained high through the 22 hours of the study. The values declined after an initial rise in control rats (Field and Smith, 1964). Comparison of magnesium-deficient and control rats given 28Mg before sacrifice at intervals up to 48 hours, showed rapid uptake (at 2-4 hours) that was most marked in liver, heart, and kidney (Chutkow, 1965). All subcellular fractions of the myocardium, from rats kept magnesium deficient for 32 days, exhibited avidity for 28Mg that had been given 12 hours before sacrifice (Ryan et al., 1973). Rats that had been repleted for 18 days before the 28Mg injection did not show greater than control 28Mg uptake, indicating repair of the myocardial deficit within that period of time.
7.2. The Magnesium Status of the Myocardium
The amount of myocardial magnesium might be the factor that determines cardiac response to the many cardiopathic factors in our environment. Dietary imbalances that increase magnesium requirements at the same time that less is ingested have been shown to lower myocardial magnesium levels. In experimental animals, such short- or long-term magnesium deficiencies have produced arrhythmias, coronary arterial lesions, and light- and electron-microscopic evidence of damage that is intensified by stress. Hormones that stress causes to be secreted (e.g., catecholamines and corticosteroids), and drugs or hormones that cause further loss of magnesium, particularly when associated with retention of calcium (e.g., diuretics, digitalis, vitamin D, dihydrotachysterol), have similar effects.
This brings us to the concept of "pluricausal cardiomyopathy," a term used by Selye (1961, 1969) and Raab (1969, 1972) as preferable to the limiting term "coronary heart disease." They used it to encompass also hormonal and dysionic responses to emotional, as well as drug-induced stresses and metabolic aberrations. Selye (1969) commented that deficiencies in dietary potassium, magnesium, or chloride each predisposes to cardiac necrosis closely resembling that of his electrolyte-steroid cardiac-necrosis (ESCN) experimental model in that all produce extensive, usually multifocal myocardial necrosis. Excessive concentrations of epinephrine like substances in the heart of a young athlete who had died suddenly (Raab, 1943a), and in hearts of patients who had died with angina pectoris and other cardiac dysfunctions (Raab, 1943b), and the similarity of the ECG changes of patients with IHD to those of animals or humans given epinephrine, led Raab to consider stress-induced hormonal (catecholamine and corticosteroid) excess as basic to the disorder he termed cardiac "dysionism" (Raab, 1972). He observed that major shifts in myocardial electrolytes can lead to disturbances in cardiac rhythm, contractility, structure, and ultimately to cell necrosis. His emphasis was on the depletion of intracellular potassium, but he observed that this was usually paralleled by loss of glycogen and magnesium and by entry of sodium into the myocardial cells.
Since experimental magnesium deficiency was first recognized as causing cardiac damage, both functional and morphological, and since development of the electron microscope has permitted demonstration of mitochondrial changes (remarkably similar to those produced by experimental ischemia) that can explain the dysionism referred to by Raab (1969, 1972), the cardiac changes caused by magnesium deficiency are presented before the discussion of the role of magnesium loss in dysrhythmias.
7.3. Myocardial Changes with Magnesium Deficiency or Loss-(Animal)
7.3.1. Experimental Magnesium Deficiency
Functional and histologic abnormalities of the heart were demonstrated in magnesium-deficient rodents and ruminants over 40 years ago (Greenberg et al., 1936; Moore et al., 1936) and low cardiac magnesium levels in the failing human heart even earlier (Wilkins and Cullen, 1933). The nature of the damage that is caused by experimental magnesium deficiency, and the protective effects of magnesium administration, have been demonstrated in many animal models of cardiovascular disease (Reviews: Selye, l958g; Bajusz, 1965; Heggtveit, 1965c, Raab, 1969; Lehr, 1969; Rigo, 1971; Rotman, 1971; Szelenyi, 1971; Seelig, 1972; Seelig and Heggtveit, 1974; Seelig and Haddy, 1976/1980). As indicated, the "pure" magnesium deficient heart has histological myocardial lesions that are predominantly perivascular (around the damaged small coronary arteries) and thus probably reflect hypoxia secondary to the early arterial damage.
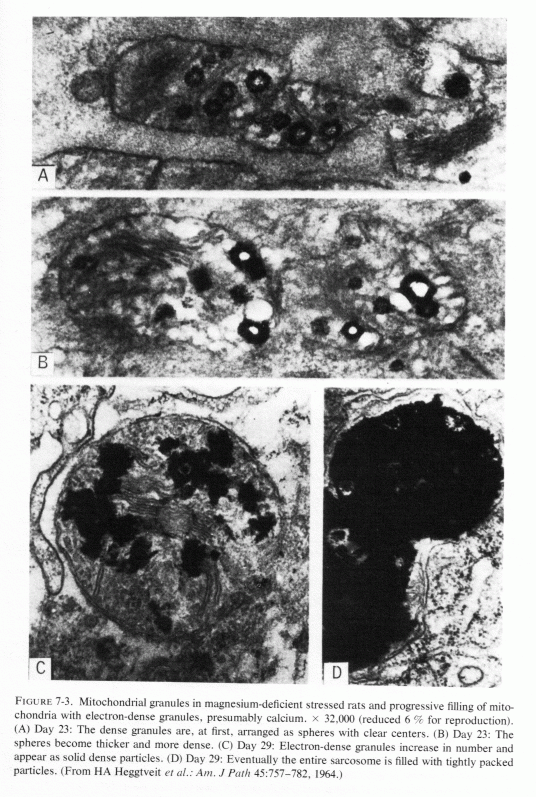
Light microscopic lesions (including focal myocardial necrosis, exudative inflammation, and varying degrees of calcification and collagen deposition) were seen in rats that were magnesium depleted for 14-36 days, the degree of damage being directly related to the duration of the depletion (Heggtveit et al., 1964; Heggtveit, 1965b,c). A group that was also cold stressed (swimming in ice-water bath for four minutes) twice daily the last two days before sacrifice exhibited the most severe damage; they were the only rats to exhibit grossly evident cardiac damage. Many of the myocardial lesions were perivascular, surrounding small ramifications of the coronary arteries, but this was not a consistent finding. Primary arterial damage, other than edema of the endothelium, was not noted. Ultrastructural changes in the myocardium were most pronounced in and around the areas of necrosis. Like the magnesium-deficient rats reported by Nakamura et al. (1961) that had swollen mitochondria after 12 days of magnesium deficiency, those of Heggtveit et al. (1964) also showed mitochondrial or sarcosomal swelling and distortion (at 14 days). There was vacuolization of enlarged sarcosomes, clumping of cristae, and progressive deposition of electron-dense material, which eventually filled the entire saracosome or mitochondrion especially in the magnesium-deficient stressed rats (Fig. 7-3). Rats given the same diet, but with magnesium supplements, developed no cardiac lesions, whether or not they were cold stressed.
Fragmentation and loss of myofilaments (which make up the myofibrils) both accompany and follow the sarcosomal changes. Thus, there is disruption of "Z" bands and "M" lines, with spaces within the myofibers. Aggregating within these spaces (corresponding to vacuoles seen by tight microscopy) are dilated components of the sarcoplasmic reticulum, damaged sarcosomes and ground substance, lipid droplets and glycogen particles. Finally, the sarcolemmal membrane ruptures or disappears, and the altered sarcoplasmic constituents spill into the interstitial space, where they are ingested by macrophages aligned alongside necrotic muscle cells (Heggtveit, 1965c).
Mishra (1960b), who had found that the mitochondrial fraction of hearts from magnesium-deficient rats was diminished, reasoned that such a loss, which is linked to oxidative phosphorylation, might be responsible for defective ability of magnesium-deficient mitochondria to maintain ionic gradients and for metabolic and respiratory cell injury leading to myocardial necrosis. DiGiorgio et al. (1962) proposed that since the amount of magnesium in the distorted cardiac sarcosomes was the same (or even more) in the magnesium-deficient than in the control rats, possibly it was in a form unsuitable for coupling of oxidation to phosphorylation.
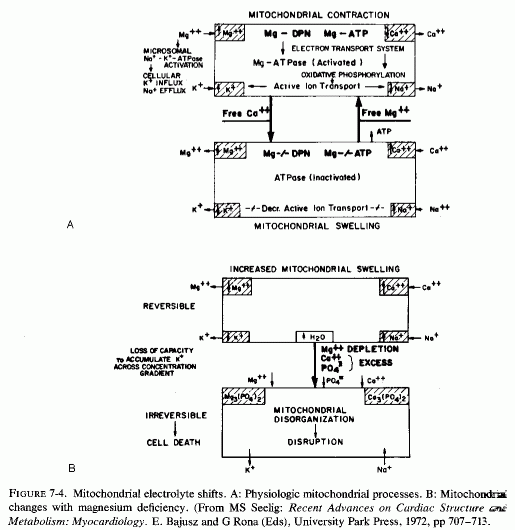
Ultramicroscopy has shown that magnesium deficiency for as little as 12-14 days has caused cardiac mitochondria to swell (Nakamura et al., 1961; Heggtveit et al., 1964; Heggtveit, 1965b,c); that such swelling is not physiologic, such as occurs during ionic flux (Fig. 7-4A), but is pathologic (Fig. 7-4B). It is associated with mitochondrial disruption and disorganization. The electron dense particles probably consist of calcium (e.g., as phosphate crystals). Possibly some of the mitochondrial magnesium is similarly made unavailable (Jennings, 1969; Seelig, 1972). Such redistribution of the calcium and magnesium ions, taking them and the inorganic phosphate out of the pool available for oxidative phosphorylation, might be contributory to irreversible mitochondrial damage. It must be noted that the mitochondria from a magnesium-deficient rat that had marked mitochondrial and sarcosomal calcium granular deposition (Heggtveit et al.., 1964; Heggtveit, 1965b,c) were from a rat that was cold stressed.
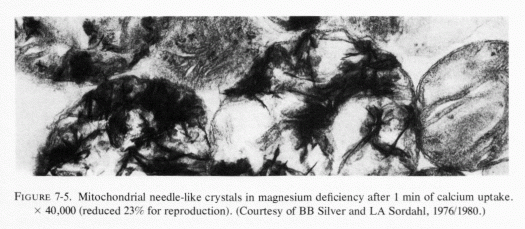
Heggtveit (1965c) has reviewed the data correlating the close interdependence between mitochondrial structure and function and has observed that early sarcosomal alterations are fundamental to the evolution of the cardiac necrosis of magnesium deficiency (Heggtveit et al., 1964). He noted that the calcium accumulation occurring in magnesium deficiency begins before the cell dies. A recent in vitro study provides evidence that magnesium modulates calcium uptake in cardiac mitochondria (Silver and Sordahl, 1976/1980). Respiration-supported calcium uptake by rabbit-heart mitochondria in magnesium-free medium was almost double that in the presence of magnesium. Furthermore, in the absence of magnesium, the calcium crystals in the mitochondrial matrix were needlelike. On addition of magnesium to previously magnesium-free suspensions, they underwent transformation into an apparently destructive granular type with dendritic crystals that obliterated the internal mitochondrial structure (Fig. 7-5). In the presence of magnesium, spheroidal-amorphous calcium crystals form in the mitochondria. Silver and Sordahl (1976/1980) suggest that the magnesium modulation of the calcium uptake, and its influence on the shape of the crystals, is consistent with the protection afforded by magnesium against the necrotizing effects of calcium on myocardial cells when magnesium levels are low (Janke et al., 1975; Lehr et al., 1975).
7.3.2. Magnesium Loss from the Hypoxic Heart
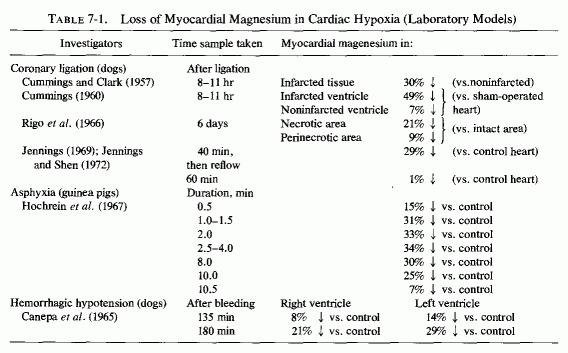
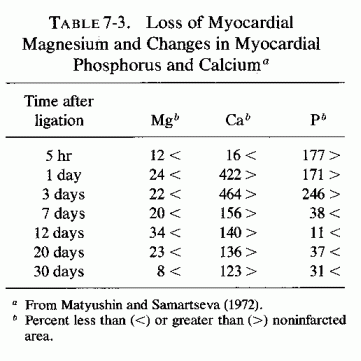
There is loss of myocardial magnesium from hearts of experimental animals with coronary ligations after asphyxia (Table 7-1) and from infarcted areas of human hearts (Table 7-2). Why the drop in myocardial magnesium was greater after transient ischemia (following reestablishment of circulation) than it was in dog hearts with permanent ischemia (Jennings and Shen, 1970/1972) was not explained, but it is in accord with the short-term (up to 10 minutes) findings of Hochrein et al. (1967) with asphyxiated guinea pigs and with the long-term study of electrolyte changes in infarcted versus noninfarcted areas of the heart from 5 hours at intervals to 30 days (Table 7-3, Matyushin and Samartseva, 1972). It is conceivable that the short-term fall in magnesium, reaching almost normal levels 2½ minutes after cardiac arrest, might reflect formation of unavailable (possibly phosphate precipitate) magnesium within the mitochondria (Seelig, 1972).
Jennings et al. (1965,1969) have shown that myocardial cells show some mitochondrial damage (some loss of cristae and matrices), some loss of glycogen and some margination of chromatin material within 15 minutes after coronary occlusion. Restoration of blood flow by no more than 18 minutes allowed for resumption of normal structure and function (back to aerobic from anaerobic metabolism). Longer periods of ischemia resulted in irreversible mitochondrial damage, with loss of cristae, disruption of limiting membranes, and intramitochondrial granules (after 40 minutes of ischemia). The dead or dying cells exhibit loss of magnesium, potassium, and acid-soluble phosphate, and gain of sodium, chloride, and water, an electrolyte distribution similar to that of extracellular fluid (Jennings et al., 1969, 1970). Later, the calcium and phosphorus levels rise (Jennings and Shen, 1970/1972; Shen and Jennings, 1972), probably as calcium phosphate granules form. Jennings (1969) has suggested that crystallization or binding of essential co-factors such as phosphate, calcium, and possibly magnesium in the granules might contribute to irreversible mitochondrial failure. A. Schwartz (1971/1972) commented that mitochondria have the ability to sequester large amounts of calcium, and that if enough calcium interacts with the mitochondrial membranes, there is significant uncoupling of oxidative phosphorylation. Shen and Jennings (1972) demonstrated that ischemic injury causes abnormal calcium uptake as dense intramitochondrial granules, which are an important feature of irreversible cellular injury.
The later, lesser rises in myocardial magnesium that occur in hypoxic hearts probably must be otherwise explained than by accumulation of magnesium phosphate crystals or granules in the mitochondria. Page et al. (1972) have shown that myofibrillar magnesium and mass increases, and the ratio of mitochondrial volume to cell volume decreases in rabbit hearts with mechanical interference with left ventricular outflow, If such myofibrillar sequestration of magnesium occurs in the surviving cells in the area in which ischemia has been induced, perhaps this explains at least partially the later rise in myocardial magnesium levels.
Heggtveit (1965c, 1969) and Heggtveit and Nadkarni (1971), in their reviews of electron microscopic findings of myocardial ischemia, considered the similarities in mitochondrial changes to those of magnesium depletion and catecholamine cardiopathy, which Lehr and his associates had correlated with early loss of myocardial magnesium and accumulation of calcium (Lehr et al., 1966; Lehr, 1969). Heggtveit (1969) pointed out, however, that early nuclear changes are characteristic of ischemic injury, whereas nuclear chromatin clumping occurs only late, after severe sarcoplasmic damage of magnesium deficiency. He commented that correlation of ultrastructural data with biochemical findings confirms the importance of catecholamine release and ionic shifts (early loss of magnesium, potassium, and phosphate with influx of calcium, sodium, and water) in the early evolution of ischemic myocardial damage. Poche (1969) reported that capillary endothelial swelling, with reduction in luminal caliber of the microcirculation, is significant in the pathogenesis of multifocal hypoxic myocardial necrosis. Such endothelial swelling has been reported in magnesium deficiency (Heggtveit 1965c; Hungerford and Bernick, 1976/1980), as have endothelial and medial proliferation. These arterial changes of "pure" magnesium deficiency, thus, might contribute to the hypoxia-like myocardial lesions seen in magnesium deficiency, and might contribute to the decreased resistance of the myocardium to stress factors, such as Heggtveit (1969) suggested might "condition" a chronically ischemic heart to severe response to subsequent acute episodes.
7.3.3. Magnesium Loss from the Stressed Heart or in Association with Catecholamine Administration
Raab (1943b, 1966, 1969) was the first to point out that catecholamines increase cardiac work and oxygen consumption to the extent that relative hypoxia is produced, particularly in the presence of coronary disease that prevents adequate oxygenation. Relative cardiac hypoxia is also produced with cardiac overload (Hochrein and Lossnitzer, 1969) with similar consequences: stress-induced dysionic status in the myocardium, which leads to functional and finally structural abnormalities in the heart that can result in sudden death from arrhythmias, cardiomyopathies that can lead to chronic heart disease, or the more widely recognized "coronary heart disease." Although he placed major emphasis on the loss of potassium from myocardial cells, he observed that rats stressed by isolation also had low myocardial magnesium levels (Raab et al., 1968), and that patients who had died with ischemic heart disease also had low myocardial magnesium levels (Raab, 1969). He considered the catecholamine release a major mediating cardiopathic response to stress, but called attention to evidence that catecholamine mobilization of free fatty acids from adipose tissues is dependent on the presence of glucocorticoids (Maickel et al., 1966). Thus, he considered the stress release of catecholamines and corticosteroids additive in cardiopathic potential.
It is thus particularly unfortunate that acute coronary occlusion is a very stressful event that stimulates secretion of both of the major groups of adrenal hormones, those of the cortex and the medulla. As regards the catecholamines, such secretion takes place, not only from the adrenal medulla, but also within the heart itself, which synthesizes, stores, and releases norepinephrine (Raab and Gigee, 1955; Braunwald et al., 1964).
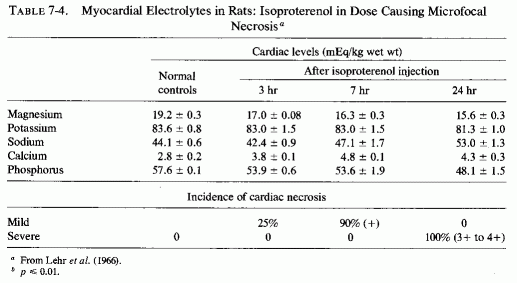
Much work has been done on the nature of the gross, histological, and electron- microscopic myocardial necrosis produced by high doses of the potent β-adrenergic amine, isoproterenol, since the work of Rona et al. (1959), which was shown the same year to be intensified by mineralocorticoids (Chappel et al., 1959). Ferrans et al. (1964, 1969), using the high dose (85 mg/kg) that consistently produces large infarction-type lesions, found that mitochondrial swelling, vesiculation, and crystolysis developed early, and myofibrillar degeneration later. Zbinden and Bagdon (1963) found that even with relatively low doses, the myocardial lesions occurred regularly and were located predominantly at the interventricular septum, the apex, and the wall of the left ventricle. The location of the lesions at the sites that had been shown to have the greatest affinity for 28Mg (Glaser and Brandt, 1959; Glaser and Gibbs, 1962) and to have the highest magnesium concentration (Lazzara et al., 1963; Burch et al., 1965), and the similarity of the ultramicroscopic lesions to those produced by magnesium depletion are inferential evidence that the catecholamine-induced myocardial might be mediated by loss of myocardial magnesium. Lehr et al. (1966), using small enough doses of isoproterenol (5.25 mg/kg) to produce disseminated myocardial necrosis, rather than grossly evident necrosis, proved the first myocardial changes to be loss of magnesium and phosphorus increased calcium; sodium and potassium changes occurred later (Table 7-4). The decrease in magnesium in the myocardium was demonstrable as early as one hour after isoproterenol injection, even preceding the mitochondrial changes that were evident at two hours. In view of the importance of magnesium in oxidative phosphorylation, it is not surprising that similarly small doses of the catecholamines caused its depression in cardiac mitochondria (B. Sobel et al., 1966).
There is another magnesium/catecholamine interrelationship that should be considered. Magnesium and calcium have reciprocal effects on storage or release of catecholamines from adrenergic granules in the adrenal medulla. Mg-ATP stimulates amine incorporation in adrenal medullary granules (Carlsson et al., 1963). Calcium stimulates and magnesium inhibits release of catecholamines from the granules. (W. Douglas and Rubin, 1964; J. Burn and Gibbons, 1964). Since catecholamine granules, epinephrine, or related substances have been demonstrated in the myocardium (Raab, 1943a,b; Potter and Axelrod, 1963b), particularly in hearts from patients with angina pectoris (Raab, 1943b), the observation that in vitro addition of magnesium stabilizes the catecholamines in the heart, preventing releasing of norepinephrine, (Potter and Axe 1963a) might be significant in the clinical situation.
In his thesis on the effects of magnesium deficiency in the rat, C. Johnson (1965) showed that the adrenal medullary levels of epinephrine fell: 23% decrease after 8 days of deficiency and 46% decrease after 12 days of deficiency. Possibly this reflects increased release of epinephrine from the adrenal medullary granules in magnesium deficiency. The same rats also exhibited a slight increase in myocardial catecholamine levels that was associated with low cardiac magnesium and ATP levels.
7.3.4. Corticosteroid + Phosphate-Induced Myocardial Necrosis
In addition to the increased output of catecholamines in response to stress, the secretion of corticosteroid hormones is also increased. Selye approached the problem of cardiovascular disease associated with stress from the standpoint that mineralocorticoid secretion was predominantly to blame, particularly in subjects with dietary excesses of sodium, phosphate, and sulfate (Review: Selye, 1958f). In the historical introduction to his 1958 monograph, Selye referred to the early work on the importance of ionic interactions for the function of cardiac muscle in vitro, which led to the recognition of the advantages of physiologically balanced perfusion electrolyte solutions over saline. He reviewed the discovery that had been made at the turn of the century of "acute interstitial myocarditis" for which no cause was identifiable, and commented on the similarity of those lesions to those discovered about the same time (1904) to be produced by experimental overdosage with cardiac glycosides. When irradiated ergosterol preparations became available in 1929, he found that intoxication with sterols of the vitamin D group also produces generalized arterial calcification and focal myocardial necrosis and calcification (Selye, 1929). He noted that all of these myocardial disorders, including that caused by potassium deficiency (Schrader et al., 1937) were characterized by focal necrosis and by inflammatory infiltration (similar to that reported by others using magnesium-deficient diets, supra vide). Then he found that multiple doses of the mineralocorticoid desoxycorticosterone (DOC) caused minute myocardial necrosis in rats, an effect attributed to loss of potassium (Darrow and Miller, 1942). Chicks, fed a ration that was rich in sodium chloride, were more susceptible to DOC-cardiotoxicity (and nephrotoxicity) (Selye and Stone, 1943), and sodium chloride aggravated myocardial necrosis caused by potassium deficiency (Cannon et al., 1953). Selye (1958f) noted that in the "control" potassium-deficient rats in the latter studies, the chloride salt of magnesium had been used as a "filler," in place of the sodium salt, magnesium's protective role not then being generally known. Development of the electrolyte (sodium phosphate)-steroid (mineralocorticoid)-cardiac necrosis (ESCN) model permitted demonstration of some of the factors that intensified or mimicked the myocardial, noninfarctoid lesions. It also permitted investigation of factors with cardioprotective properties (Selye, 1958f). Sodium salts of phosphate and sulfate intensified the lesions; magnesium and potassium chlorides were protective (Selye, 1958 a,d,g, 1961, 1969, 1970b; Selye and Mishra, 1958; Selye and Gabbiani, 1965). Because the ESCN-like lesions could be produced by unrelated agents-cardiac glycosides, vitamin D derivatives, epinephrine, stress-as well as by deficiencies of magnesium or potassium or both, Selye postulated that there must be a common pathway. Also noted was the efficacy of chloride salts of magnesium and potassium against many cardionecrotizing agents (Bajusz and Selye, 1960a). In his surveys of the evolution of the concept that stress contributes to cardiovascular diseases, Selye (1961, 1970a,b) described experiments that, depending on conditioning factors, including stress and the "stress hormones" (ACTH. corticosteroids, and the catecholamines), can produce or prevent cardiovascular lesions. He investigated the importance of mineralocorticoids in the pathogenesis of hypertension, edema, and myocardial lesions (in animals and in human disease) and showed that glucocorticoids that lack a significant mineralocorticoid component do not intensify the ESCN. It is noteworthy that chronic hypercorticism that is associated with sodium and water retention is associated with renal loss of both potassium and magnesium (Review: Massry and Coburn, 1973), and thus functions to increase levels of the "conditioning" cation, sodium, while causing loss of the "protective" cations, potassium and magnesium.
That such a combination of responses can have serious consequences is indicated by the extraordinary potentiation of acute isoproterenol-cardiotoxicity by pretreatment with DOCA and saline (Guideri et al., 1971, 1974, 1978). Such conditioning of the rats resulted in death from fibrillation within 15 to 30 minutes after 150 µg/kg to 0.1 mg/kg of isoproterenol subcutaneously, an amount far below the minimally toxic dose (5 mg/kg) used to produce microfocal necrosis in earlier studies of isoproterenol alone (Lehr et al., 1966; Lehr, 1969) or in combination with mineralocorticoids (Chappel et al., 1959). The myocardial electrolyte pattern showed significant accumulation of sodium and loss of potassium, magnesium, and phosphate. As for the glucocorticoids, the secretion of which is also increased in stress, they further contribute to the metabolic abnormalities by the dependence of catecholamine metabolization of free fatty acids on their presence (Maickel et al., 1966).
Catecholamine levels were not measured in the magnesium-deficient rats (dietary magnesium: 12 mg/g diet for seven weeks) that had much greater degrees of cardiac necrosis when cold stressed than did control cold-stressed rats fed a standard magnesium-supplemented deficient diet (Mishra, 1960e; Heroux et al., 1971/1973). In the latter series (Heroux et al.), four of the eight stressed rats also had 1+ to 2+ cardiac damage (compared to 1+ to 3+ cardiac lesions in all seven of the magnesium-deficient rats). The investigators considered the possibility that the control diet might have been suboptimal in magnesium, and that the control rats might resemble humans on suboptimal magnesium intakes in their susceptibility to myocardial damage of stress.
7.3.5. Hereditary Cardiomyopathy of Hamsters
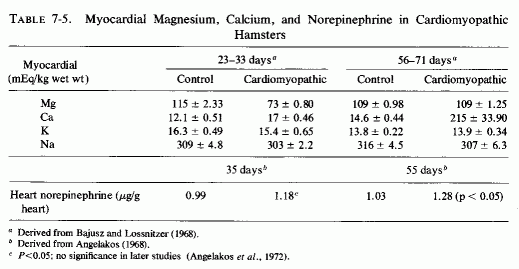
An interesting model of genetic cardiomyopathy, developed in dystrophic hamsters, might help in elucidating some of the myocardial interrelationships with magnesium and catecholamines. These hamsters, that consistently develop focal myocardial degeneration and myolysis between the 30th and 40th days of life, exhibit decreased magnesium and increased calcium in their myocardium even before the lesions develop (Bajusz and Lossnitzer, 1968), and increased levels of cardiac norepinephrine not long thereafter (Angelakos, 1968; Table 7-5). The low magnesium levels did not persist, but the calcium levels rose markedly in the 56- to 71-day-old cardiomyopathic hamsters, at a time when the norepinephrine levels had risen further (Angelakos, 1968; Angelakos et al., 1970-1972). By the time heart failure had ensued (120 days), the norepinephrine levels had dropped to half the control (young animal) levels, but to a quarter that of the same-age controls (Angelakos, 1968). Cardiac catecholamine stores also decrease once heart failure develops in other experimental models and in human heart disease (Angelakos et al., 1969).
7.3.6. Stress and Free Fatty Acids/Myocardial Necrosis and Magnesium
That catecholamines exert a lipolytic effect and increase circulating free fatty acids has been considered earlier. The significance of this on the response of the heart can be considerable. Balazs et al. (1962) and Balazs (1972) have shown that injected catecholamines or stress-induced catecholamines secretion is much more likely to cause serious myocardial damage in obese than in normal rats. This supports the contention that catecholamine-induced lipolysis can be a significant risk factor, especially in overweight patients.
Although free fatty acids can be utilized by the myocardium as an oxidative substrate, there is growing evidence that high levels of free fatty acids (e.g., mobilized by catecholamines) are cardiotoxic (Rosenblum et al. 1965; Opie, 1969; Hoak et al., 1970-1972) and can interfere with myocardial function, especially in association with hypoxia (A. Henderson and Sonnenblick, 1970, 1970/1972; Shug and Shrago, 1973). Opie (1969) has evaluated the relative importance of glycolytic and fatty acid metabolism of the heart, and points out that with excesses of fatty acids and triglycerides, there is substantially increased cardiac oxygen consumption. This can intensify the relative myocardial hypoxia caused by stress, especially in the presence of coronary insufficiency.
The possibility that high levels of free fatty acids in the blood might contribute to symptoms of alcoholics by binding magnesium has been mentioned earlier, as has the favorable response to magnesium of hyperlipemic patients with occlusive arterial disease (Seelig and Heggtveit, 1974). It is possible that postinfarction arrhythmia might be related either to excessive catecholamine release in response to the stress of the cardiac injury, or to catecholamine-induced increase in circulating fatty acids. The catecholamines are apt to lower the cardiac magnesium levels; the free fatty acids might bind magnesium in the blood. Perhaps increased myocardial lipids, such as have been attributed to catecholamine-lipid mobilization in rats injected with sympathomimetic agent (Ferrans et al., 1964; 1969) and in the ESCN model, (Prioreschi, 1966), might be the result of inactivation by the intramyocardial fats of cellular magnesium. Direct evidence that a variety of dietary fats (corn oil, peanut oil, olive oil, pork fat, butter, and chicken fats, as well as saturated and unsaturated fatty acids) greatly increase the sensitivity of the rats to ESCN has been provided (Selye, 1961; Selye et al., 1969).
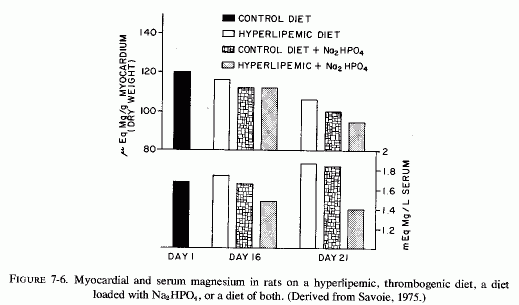
The cardiovasopathic diet developed by Sos and his co-workers that produces spontaneous myocardial infarctions includes saturated fats and hyperlipemic and hypercalcemic nutrients. Although the diet produces only minor serum electrolyte changes it substantially lowers myocardial magnesium levels. A similar diet, which was described as thrombogenic and which resembles the diet developed by Vitale and his co-workers to produce atherogenesis, incorporates propylthiouracil and cottonseed oil; when Na2HPO4 is added it causes nonocclusive infarctoid myocardial lesions (Savoie, 1972a,b, 1975). Anticholesterolemic agents lower the blood lipids, but are ineffective in protecting against the myocardial necrosis (Savoie, 1972b). The potassium-sparing agents (triamterine and spironolactone), and, to a lesser degree, potassium chloride, are partially protective against the cardiac lesions but not against the hyperlipidemia; only magnesium chloride prevented the cardiac necrosis (Savoie, 1972b). Similarly, amiloride, another potassium-sparing agent, inhibits the development of cardiac lesions produced when corn oil is added to the diets of rats on the ESCN regimen, although the protection is not complete (Kovacs et al., 1969; Solymoss et al., 1969). Ultramicroscopy showed that there was still evidence of myofibrillar damage and mitochondrial lipid droplets, although marked focal lipid accumulations were prevented (Kovacs et al., 1969). Also, the blood lipids were not normalized (Solymoss et al., 1969). Savoie (1971b) demonstrated protection against myocardial necrosis in a comparable model also for triamterene and spironolactone, the agent that blocks aldosterone under conditions of chronic hypersecretion (Massry and Coburn, 1973), such as is seen in heart disease (H. Wolff et al., 1957) and in primary aldosteronism in which it has been associated with magnesium loss (Mader and Iseri, 1955; Milne et al., 1957). All of these potassium-sparing agents also protect against the hyperlipidemic cardiac necrosis that is intensified by stress, epinephrine, or digitalis (Savoie, 1971a,b). Although amiloride did not prevent the lowering of myocardial magnesium in the ESCN+ corn-oil model that causes severe damage in a few days, and in fact actually lowered it somewhat (Solymoss et al., 1970) the potassium-sparers (amiloride and triamterene) also exert some magnesium-sparing activity (Hänze and Seybirth, 1967; Heidland et al., 1970, 1973; Walker et al., 1972). Whether this effect contributed to their partial efficacy in the hyperlipidemic +Na2HPO4 model, in which they were more effective than potassium chloride, but less effective than magnesium chloride (Savoie 1972b) should be further studied. It is of interest that in this less acute model, which possibly is more similar to the situation produced by human dietary indiscretions, the addition of sodium phosphate to the hyperlipidemic diet caused substantial lowering of both myocardial and serum magnesium levels by day 21 (Fig. 7-6, Savoie, 1975). (Note should be taken that in the hyperlipidemic and in the high phosphate-fed rats, the serum magnesium level did not reflect the drop in the myocardial levels.) Savoie (1975) considers the critical factor in the Na2HPO4 and hyperlipidemic rats to be their susceptibility to mitochondrial dysfunction caused by the low myocardial magnesium levels. Since the Na2HPO4 and the hyperlipidemia alone also lowered the myocardial magnesium levels markedly, it seems plausible that each imbalance could increase the susceptibility of the heart to stresses or other agents that further lower myocardial magnesium levels, with resultant arrhythmias or necrosis. This investigator has recently correlated the protective effect of magnesium in this model with its lowering of free cholesterol levels in the heart (Savoie and DeLorme, 1975/1980).
7.3.7. Myocardial Loss of Magnesium after Parathyroidectomy and Sodium Phosphate Load
Severe multifocal myocardial and renal necrosis, produced in parathyroidectomized rats given Na2HPO4 is preceded by markedly lowered myocardial (and renal) magnesium levels (Lehr et al., 1966; Lehr, 1969). In addition, the microcirculation of the heart is damaged; many arterioles and precapillaries show loss of the normal architecture, with the presence of arterial and periarterial PAS-positive material. After a single Na2HPO4 load, there is clumping of myocardial sarcosomes, edema, and disturbances in the normal myofibrillar pattern. After two sodium phosphate loads (at 24 hours), the mitochondria are swollen and exhibit disappearance of cristae and formation of granular debris; there is also margination of nuclear chromatin. Lehr (1969) commented that the consistent, significant early shifts in tissue cations (decreased magnesium and phosphorus, and increased sodium and calcium) in this experimental model, as well as in other very different models (e.g., catecholamines, ESCN, and cardiac overload) and the fact that the ionic shifts precede morphological damage, suggest that they are the cause, not the consequence of the myocardial damage.
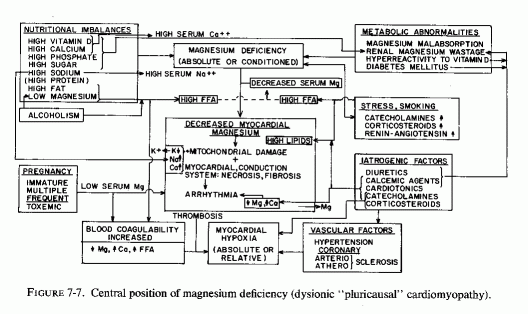
7.4. Cardiac Magnesium Loss: Central to Cardiac Dysionism, Disease, and Dysfunction (Fig. 7-7)
As indicated in the sections on the effects of magnesium deficiency on the arteries, early damage to the small coronaries with narrowing of their lumina is characteristic of magnesium deficiency. Such myocardial arterial disease is not what is referred to by "coronary disease," but it certainly contributes to microfocal areas of hypoxia, which can give rise to the microfocal necroses, infiltration, and fibrosis that have been described in magnesium-deficient animals (Review: Seelig and Haddy, 1976/1980). It is provocative that Lehr (1965b, 1969, 1972) and his co workers (Lehr et al., 1966, 1970/1972, 1976/1980), who proposed that the loss of myocardial magnesium might contribute to the disseminated myocardial necrosis caused by dissimilar agents (including catecholamines and sodium phosphate loading of corticosteroid-treated or parathyroidectomized rats), had also implicated damage to the microcirculation (Lehr, 1965a, 1966, 1969). If magnesium nutritional deficiency or drug-induced myocardial loss is a basic contributory factor [and it has been shown to predispose also to the dysionism (decreased potassium and increased sodium), as well as to increased accretions of mitochondrial calcium (Lehr, 1969, 1972; Review: Seelig, 1972)], then Lehr was correct in both postulates.
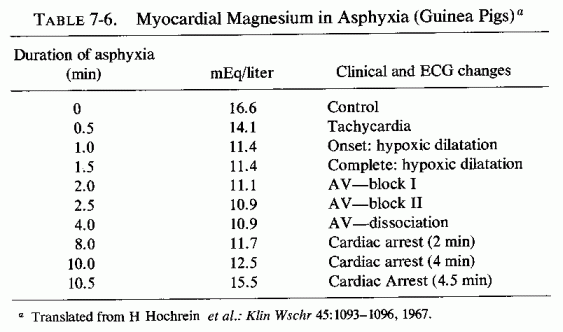
The loss of magnesium from the myocardium occurs so soon after hormonal or other challenge as to be suggested as a chemical means of detecting one of the earliest characteristics of myocardial damage (Lehr et al. 1976/1980). Its early loss after experimentally induced hypoxia (Table 7-6, from Hochrein et al., 1967) and from the heart after ischemia from coronary ligation suggests that the loss of magnesium from the fine structures of the myocardium are probably basic to the myocardial damage. Nutritional magnesium deficiency can result in early mitochondrial loss and damage directly or secondarily, as a result of focal ischemia from the narrowing of the intramyocardial vessels. Whether the hypoxia is relative or absolute, there are both increased net loss (efflux) of magnesium from myocardial cells and decreased magnesium influx during hypoxia (Polimeni and Page, 1974).
Evidence is presented that common to a wide variety of experimental models of experimental cardiomyopathy is early loss of myocardial magnesium. In his discussion of the lesions produced not only by isoproterenol, but by low doses of other catecholamines, and in other cardiopathic models [i.e., parathyroidectomy and phosphate loads, mineralocorticoid and phosphate loads (ESCN model of Selye), and hypoxic heart failure (Hochrein and Lossnitzer, 1969)], Lehr (1969) proposed that the common denominator was magnesium loss from the myocardium. He proposed that the magnesium loss is primary, and deserves closer scrutiny in view of its importance in the vital energy processes of the cell. He further proposed that its depletion might contribute to the initiation of cellular injury.
It is the premise of this section that the underlying factor-that which determines whether the individual will withstand stress or other potentially cardiopathic factors-is the adequacy of magnesium in his heart. Some might develop dysrhythmias (possibly suddenly fatal) as a result of inadequacy of the damaged magnesium-dependent mitochondrial enzyme system to maintain normal ionic equilibrium, or as the conduction system is affected or myocardial cell excitability is increased by a low magnesium/calcium ratio (infra vide). Others might develop coronary arterial disease, including hypertension, and microscopic or gross myocardial lesions that lead to chronic heart disease. The foregoing section on the lability of cardiac magnesium shows that magnesium can be readily lost from the heart, but that it can also be quickly repleted. Barnes (1962) showed that puppies kept on magnesium deficient diets for two months lost proportionally as much magnesium from the heart as they did from bone, the major magnesium store of the body. The lower myocardial magnesium levels in residents of soft-water areas than in dwellers in hard-water areas (see Chapter 1) supports the contention that long-term suboptimal magnesium intakes are associated with loss of magnesium from the heart, and with a high incidence of sudden death from IHD.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}